Data Management
General overview
Massive Parallel Sequencing (MPS) data are typically large and during the analysis they are getting even bigger. We highly recommend to work out a data management plan in advance and provide us some figures regarding expected disk space. We further encourage you to keep your home and project directories clean, organised and in agreement with scientific reproducibility. Please read the SNF guidelines.
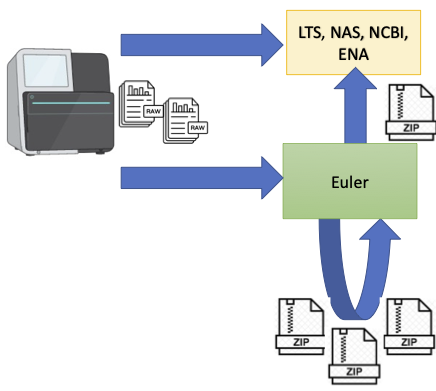
- Raw data will be transferred from the sequencing facility to Euler. At the same time we recommend to have a copy of your raw data on a long term storage (LTS) system or public databases.
- During data processing it is essential to keep only important and delete redundant files. Folders containing small files needs to be archived (zip, tar). To decide what is important and what is not can be difficult. The following Priority of different data types, the list with the general recommondations and the examples should help with data management decisions.
- After the analysis or a first step has been finished, important data should be archived and deleted on Euler. We do not recommend to use Euler as a data archive.

General recommendations
Think carefully before copying files to the GDC volume.
- Do I really need to keep these files? Data that can be reproduced quickly should be not be kept.
- Are they compressed?
- How many files do you have? If needed, archive/zip them.
- How big are the files?
- Is the transfer command specific enough? Don't just use
*to avoid copying unnecessary files.
- Work on the scratch (
${SCRATCH}or${TMPDIR}) to keep the impact on the file system as small as possible. - Generating lots or large temporary files only on the scratch.
- Do not expand archived/compressed/zipped files. Many programs can handle such files. You might also consider “on the fly” unzipping (e.g., with zcat) without writing the unzipped file(s) and folder(s) to disk. In case unzipped files are needed please use your scratch (
gunzip -c sample.fq.gz > ${SCRATCH}/<USER>). - Delete redundant data constantly (e.g. trimmed fastq file, concatenated and single files).
- Files that you like to keep on GDC home ore GDC projects needs to be compressed (vcf.gz, fasta.gz, cram).
- If you like to keep many small files, concatenate or archive them (
tar cfzv folder.tar.gz folder). - If you plan to copy files to GDC home or GDC projects make sure there is enough disk space available and inform us in case of 5 Tb data.
Number of inodes
To keep our GDC volume responsive, do not use no more than 13,000 inodes (files, soft-links, directories, hidden files) per 1 Tb of disk space.
What are important data ?
You do a read mapping and produce several types of intermediate and redundant files.
- The fastq.gz-files are in
/cluster/work/gdc/shared/<p999>. - Copy the fastq files to your scratch (
/cluster/scratch/<USER>) - Use the local scratch ${TMPDIR} for the intermediary files (e.g. sam, unsorted bam files). Output the final cram file to your scratch.
- Copy only the final cram-files to
/cluster/work/gdc/shared/<p999>
You like to to a SNP calling using GATK
- Copy the cram files to your scratch (
/cluster/scratch/<USER>). - Convert the cram to bam file or merge them.
- Run GATK on the scratch and output the vcf files to the scratch first.
- Merge the vcf files in order to keep the inodes small.
- Copy only the final vcf.gz file to
/cluster/work/gdc/people/<USER>
You like to do a lot of simulations.
- For generating 1,000,000 of small files use your scratch (
/cluster/scratch/<USER>). - You then generate summary statistics and reduce the number of files to 50,000, which you like to keep for longer. Compress the entire folder using
tar -zcvf simulations.tar.gz simulations. - Copy the archive (archive.tar.gz) to
/cluster/work/gdc/people/<USER>.
Backup (disaster recovery)
GDC home and GDC projects is backuped (1 per week) if you have deleted important data, please contact us as fast as possible. We need to know what exactly you need and can ask the Cluster Support to get it back.
Data sharing
Data up to 100 Gb you can be shared using SWITCHfilesender, Swisstransfer or polybox.
Public databases
European Nucleotide Archive (ENA)
The European Nucleotide Archive (ENA) provides different ways to upload your raw sequencing data. We recommend you either use the browser based Webin submission tool or the command line. For more details please consult ENA-docs.
Always keep a local copy of the uploaded files.
No matter wich way you go your first need a submission account.
A Open a new project in the web interface
B Prepare your data in your ${SCRATCH}.
Files that are in a human-readable text format (such as FASTQ or FASTA) must be compressed before they are uploaded to the ENA FTP server. Your raw files should already be gzipped (file.fq.gz).
If you want to combine multiple fastq files, use cat A_L1.R1.fq.gz A_L2.R1.fq.gz > A.1.fq.gz, there is no need to uncompress and re-compress the files.
You must provide the MD5 value for your files to confirm data transfer. MD5 is a function which can be applied to a file to create a 32 character string. This string is unique to the file and functions like a fingerprint: if the contents of the file change in any way the MD5 checksum will change as well.
Short Read Archive (NCBI)
The MD5sums are not required when you submit the data to SRA.
for i in *fq.gz
do
md5sum $i > ${i}.md5
done
C Upload your data (either C1 or C2; C2 is normally much faster)
C1 Webin
The ENA Webin submission service guides the user through a sequence of forms and checklists allowing interactive submission of sequence data and descriptive information.
Webin submitters are encourage to contact datasubs@ebi.ac.uk for expert advice.
C2 LFTP Command Line Client
Data upload
- Use the login node to submit the files to avoid problems with the proxy server.
- Make batches to submit your data using for example regex (A*).
- If you are going to use a submission script (not recommended) load the proxy Module.
lftp
open webin.ebi.ac.uk
user username
mput A*gz
put A*md5
# Help:
# Type `ls` command to check the content of your drop box.
# Use `mput` command to upload files.
# Use `bye` command to exit the ftp client.
D Link submitted data with meta data in the web interface.
- Register a new study if needed.
- Register samples using the appropriate checklist (minimum requirements: sample_alias; tax_id; scientific_name; common_name; sample_title; location; sampling date).
- Submit reads using the appropriate spreadsheet.
- After a couple of days verify if all samples could be successfully archived in "Run Processing Report".
- Delete data on Euler.