Job Submission
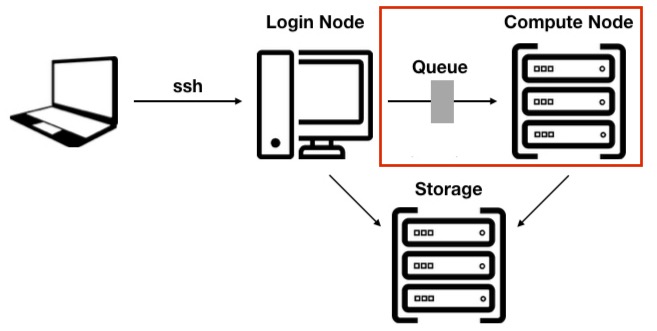
The main difference between an HPC cluster and a traditional server or workstation is that an HPC cluster is designed to break down entire pipelines into smaller units so that each step can be optimised and controlled more effectively.
Learning Objectives
- Knowing how to set up an workflow on Euler.
- Knowing how to submit a single job.
- Knowing what is an interactive node and how you can submit a job arrray.
Job submission
Let's have a look at this process.
graph LR
Input -- tool1, tool2 --> outputThe first step is to split the job in 2 sub processes.
graph LR
input -- tool1 --> tmp_out graph LR
tmp_out-- tool2 --> outputOn a server or workstation, we would run the two commands in sequence.
tool1 input > tmp_out
tool2 tmp_out > output
On a HCP cluster we will have two independent jobs and we need to give the "command" to the scheduler slurm in our case. To do this in a reproducible way, we will use submission scripts.
Below are the minimum requirements for such a submission script for step 1.
#!/bin/bash
#SBATCH --job-name=tool1 #Name of the job
#SBATCH --ntasks=1 #Requesting 1 node (is always 1)
#SBATCH --cpus-per-task=1 #Requesting 1 CPU
#SBATCH --mem-per-cpu=1G #Requesting 1 Gb memory per core
#SBATCH --time=1:00:00 #Requesting 1 hours running time
#SBATCH --output tool1.log #Log
tool1 input > tmp_out
Let's put these lines into a text file (submit_tool1.slurm.sh), make it executable and submit the command as follows:
sbatch < submit_tool1.slurm.sh
Create a submission script for the second step.
#!/bin/bash
#SBATCH --job-name=tool2 #Name of the job
#SBATCH --ntasks=1 #Requesting 1 node (is always 1)
#SBATCH --cpus-per-task=1 #Requesting 1 CPU
#SBATCH --mem-per-cpu=1G #Requesting 1 Gb memory per core
#SBATCH --time=1:00:00 #Requesting 1 hours running time
#SBATCH --output tool2.log #Log
tool2 tmp_out > output
And then submit it as follows
sbatch < submit_tool2.slurm.sh
Workflow
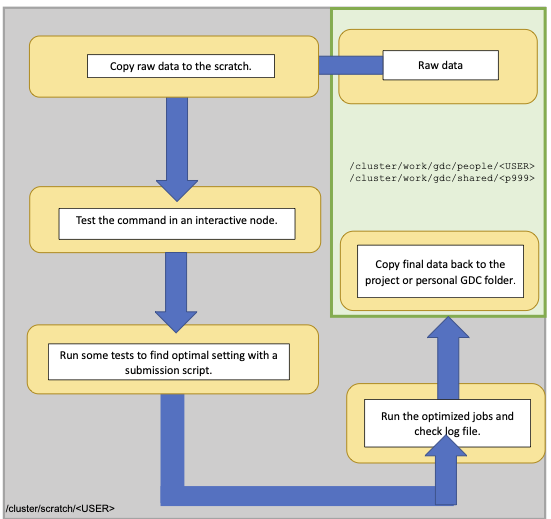
This is a typical workflow on Euler. Your working directory is the scratch, the raw data is stored on projects, work volume. We first copy the files to the scratch. In an interactive node you can then test the command. In order to do reproducible research the next step you run some test runs using a submission script to find the optimal resources (CPU, memory and time). Information about running jobs can be obtained through the monitoring tools, as explained later. Afterwards you can run all jobs, check the log file and transfer only the output that you like to keep to your project folder.
We are going to do a SNP calling. For more information on the workflow, see the SNP challenge.
Interactive node
If you like to work on a cluster like a normal server and get the outputs directly in the terminal e.g. grep some patterns or you like to test a command, use an interactive node.
srun --cpus-per-task=1 --time=04:00:00 --pty bash
Let's generate a folder on the scratch and copy the the bam files
mkdir ${SCRATCH}/mapping
mkdir ${SCRATCH}/Ref
cp /cluster/work/gdc/shared/p9999/mapping/*bam* ${SCRATCH}/mapping
cp /cluster/work/gdc/shared/p9999/Ref/* ${SCRATCH}/Ref
cd ${SCRATCH}
We first need a list with all bam files that we like to include in the analysis.
ls -1 mapping/*.sort.Q20.nodup.bam > bam.lst
Then we need a list with all chromosome names. We can use for example samtools to extract the fasta names of the reference.
module load samtools/1.17
samtools faidx Ref/ref.fasta | cut -f1 > chrom.lst
Single job
For reproducible research it is important to use scripts rather than just commands. We now going to do a joint SNP calling using bcftools.
#!/bin/bash
#SBATCH --job-name=bcf #Name of the job
#SBATCH --ntasks=1 #Requesting 1 node (is always 1)
#SBATCH --cpus-per-task=1 #Requesting 1 CPU
#SBATCH --mem-per-cpu=1G #Requesting 4 Gb memory per core
#SBATCH --time=4:00:00 #Requesting 4 hours running time
#SBATCH --output bcf.log #Log
#Load the needed modules
module load bcftools/1.16
#define in and outputs
OUT=SNPs
if [ ! -e ${OUT} ] ; then mkdir ${OUT} ; fi
REF=Ref/Ref.fasta
#The bcftools command
bcftools mpileup -f ${REF} --skip-indels -b bam.lst -a 'FORMAT/AD,FORMAT/DP' | \
bcftools call -mv -Ob -o ${OUT}/raw.bcf
The script (name: submit.bcf.slurm.sh) that you would prepare in an editor includes some explanations. The actual bcftools command is on the last line. This is the command you would run on the command-line of a regular server.
To run this submission script, you would type:
sbatch < submit.bcf.slurm.sh
Job array
Many computing tasks can be split up into smaller tasks that can run in parallel (e.g BLAST, read mapping, SNP-calling). A cluster is extremely well suited for such jobs and the sbatch command has an option specifically for this.
Let's do the SNP calling on the 20 chromosmes in parallel.
#!/bin/bash
#SBATCH --job-name=bcf #Name of the job
#SBATCH --array=1-20%10 #Array with 20 Jobs, always 10 running in parallel
#SBATCH --ntasks=1 #Requesting 1 node for each job (always 1)
#SBATCH --cpus-per-task=1 #Requesting 1 CPU for each job, 10 CPUs in total
#SBATCH --mem-per-cpu=1G #Requesting 1 Gb memory per core
#SBATCH --time=4:00:00 #4 hours run-time per job
#SBATCH --output=bcf_%a.log #log files
#Load the needed modules
module load bcftools/1.16
OUT=SNPs
if [ ! -e ${OUT} ] ; then mkdir ${OUT} ; fi
#define input and outputs
##The internal variable of slurm (1-20 in our case; see header slurm) can be used to extract the names of the chromosomes.
CHR=$(sed -n ${SLURM_ARRAY_TASK_ID}p chrom.lst)
REF=Ref/Ref.fasta
##Bcftools command
bcftools mpileup -f ${REF} --skip-indels -b bam.lst -r ${CHR} -a 'FORMAT/AD,FORMAT/DP' | \
bcftools call -mv -Ob -o ${OUT}/raw.${CHR}.bcf
To run this submission script, you would type:
sbatch < submit.bcf_array.slurm.sh
R scripts
Running Rstudio on HPC clusters can be sometimes a bit annoying, especially if you like to visualize your data. If you have entire workflows you can submit Rscripts via sbatch, save the data as Rdata and do e.g. the plotting or the filtering locally on your computer. Here an example of an R script that you use for just read a huge data file and save it as a Rdata object.
Let's copy the lines below in a file and call it Reformat.R.
#!/usr/bin/env Rscript
## Use this argument to provide file names in the command below, order must be consistent
args <- commandArgs(trailingOnly=TRUE)
library(tidyverse)
## Read table
samples <- read_csv(args[1], header = FALSE)
## Save it as a RData file
name<- paste(args[2], "RData")
save(samples, name)
As we normally don't want to load user specific data we use --vanilla option.
sbatch --ntasks=1 --cpus-per-task=1 --time 4:00:00 --wrap="Rscript --vanilla Reformat.R Fst_chromosomes1.txt Fst_chromosomes1_reduced"
This command will then output the file Fst_chromosomes1_reduced.RData
Challanges
You like to do a mapping script.The details are described in this challenge.
How you set up your mapping workflow?
We going to copy all the fastq files as well as the reference to the scratch. We might need to index the reference and make a sample list. We will split the process in two parts mapping and then filtering of the bam files.
Let's create first a single sample mapping script
#!/bin/bash
#SBATCH --job-name=mapA #Name of the job
#SBATCH --ntasks=1 #Requesting 1 node (is always 1)
#SBATCH --cpus-per-task=2 #Requesting 2 CPU
#SBATCH --mem-per-cpu=2G #Requesting 2 Gb memory per core
#SBATCH --output mapA.log #log
#Load the needed modules
module load samtools/1.16 bwa/0.7.17
##define variable
REF=Ref.fasta
CPU=${SLURM_CPUS_ON_NODE}
SAMPLE=A1
bwa mem -t ${CPU} ${REF} -R "@RG\tID:${SAMPLE}\tSM:${SAMPLE}\tPL:Illumina" ${SAMPLE}_1.fq.gz ${SAMPLE}_2.fq.gz > ${SAMPLE}.sam
#!/bin/bash
#SBATCH --job-name=mapB #Name of the job
#SBATCH --ntasks=1 #Requesting 1 node (is always 1)
#SBATCH --cpus-per-task=1 #Requesting 1 CPU
#SBATCH --mem-per-cpu=3G #Requesting 3 Gb memory per core
#SBATCH --output mapB.log #log
#Load the needed modules
module load samtools/1.17
##define variable
SAMPLE=A1
samtools sort ${SAMPLE}.sam -o ${SAMPLE}_sort.bam
samtools index ${SAMPLE}_sort.bam
samtools flagstat ${SAMPLE}.sort.bam > ${SAMPLE}_sort.stats
samtools view -hb -q 20 -F 0x800 -F 0x100 ${SAMPLE}_sort.bam > ${SAMPLE}_sort_Q20.bam
samtools index ${SAMPLE}_sort_Q20.bam
samtools collate -O -u ${SAMPLE}_sort_Q20.bam | samtools fixmate -m -u - - | samtools sort -u - | samtools markdup -r -f dup_${SAMPLE} - ${SAMPLE}_sort_Q20_fix_nodup.bam
samtools index ${SAMPLE}_sort_Q20_fix_nodup.bam
samtools flagstat ${SAMPLE}_sort_Q20_fix_nodup.bam > ${SAMPLE}_sort_Q20_fix_nodup.stats
samtools coverage ${SAMPLE}_sort_Q20_fix_nodup.bam > ${SAMPLE}_sort_Q20_fix_nodup.cov
rm ${SAMPLE}_sort_Q20.bam* ${SAMPLE}_sort.bam* ${SAMPLE}.bam
As we have now more sample to go (e.g. 10) we create an array.
#!/bin/bash
#SBATCH --job-name=mapA #Name of the job
#SBATCH --ntasks=1 #Requesting 1 node (is always 1
#SBATCH --array=1-10%10
#SBATCH --cpus-per-task=2 #Requesting 2 CPU
#SBATCH --mem-per-cpu=2G #Requesting 2 Gb memory per core
#SBATCH --output=mapA_%a.log #Log
#Load the needed modules
module load samtools/1.17 bwa/0.7.17
##define variable
REF=Ref.fasta
CPU=${SLURM_CPUS_ON_NODE}
##extract sample names 1-10
SAMPLE=$(sed -n ${SLURM_ARRAY_TASK_ID}p sample.lst)
bwa mem -t ${CPU} ${REF} -R "@RG\tID:${SAMPLE}\tSM:${SAMPLe}\tPL:Illumina" ${SAMPLE}_1.fq.gz ${SAMPLE}_2.fq.gz > ${SAMPLE}.sam
#!/bin/bash
#SBATCH --job-name=mapB #Name of the job
#SBATCH --ntasks=1 #Requesting 1 node (is always 1
#SBATCH --array=1-10%10
#SBATCH --cpus-per-task=1 #Requesting 1 CPU
#SBATCH --mem-per-cpu=3G #Requesting 3 Gb memory per core
#SBATCH --output=mapB_%a.log #Log
#Load the needed modules
module load samtools/1.17
##extract sample names 1-10
SAMPLE=$(sed -n ${SLURM_ARRAY_TASK_ID}p sample.lst)
samtools sort ${SAMPLE}.sam -o ${SAMPLE}_sort.bam
samtools index ${SAMPLe}_sort.bam
samtools flagstat ${SAMPLE}.sort.bam > ${SAMPLE}_sort.stats
samtools view -hb -q 20 -F 0x800 -F 0x100 ${SAMPLE}_sort.bam > ${SAMPLE}_sort_Q20.bam
samtools index ${SAMPLE}_sort_Q20.bam
samtools collate -O -u ${SAMPLE}_sort_Q20.bam | samtools fixmate -m -u - - | samtools sort -u - | samtools markdup -r -f dup_${SAMPLE} - ${SAMPLE}_sort_Q20_fix_nodup.bam
samtools index ${SAMPLE}_sort_Q20_fix_nodup.bam
samtools flagstat ${SAMPLE}_sort_Q20_fix_nodup.bam > ${SAMPLE}_sort_Q20_fix_nodup.stats
samtools coverage ${SAMPLE}_sort_Q20_fix_nodup.bam > ${SAMPLE}_sort_Q20_fix_nodup.cov
rm ${SAMPLE}_sort_Q20.bam* ${SAMPLE}_sort.bam* ${SAMPLE}.bam*
All your samples are successfully completed, what are the next steps?
Archive the log files.
Copy the final bam files (*_sort_Q20_fix_nodup.bam) and the archived log files into your project folder.