There are different ways of generating metabarcode (AmpSeq) data and these change over time. It is important to understand exactly how the samples were processed, which library was used and how the data was sequenced. The better you understand these steps, the better the processing will be. Here is an overview of commonly used methods.
Illumina
Most of the amplicon sequencing project we process at the GDC are paired-end MiSeq based data sets with dual-indexing. The index (also known as barcode) is read separately and the de-muliplexing (index sorting) of the sequence reads is done by the e.g. MiSeq Control Software (MCS).
You should have 2 or 4 files per sample:
- Forward read: SampleA_R1.fq.gz
- Reverse read: SampleA_R2.fq.gz
- Index 1 (i7 index) read: SampleA_I1.fq.gz
- Index 2 (i5 index) read: SampleA_I2.fq.gz
Illumina Read Orientation
Important for data processing are the R1 and R2 reads. If a two-step PCR protocol is used, all reads (with very few exceptions) will have the same orientation. If a PCR-free ligation protocol is used, the reads are not oriented. It is essential to align the reads before clustering. The same sequence in different orientations will result in two different but redundant OTUs.
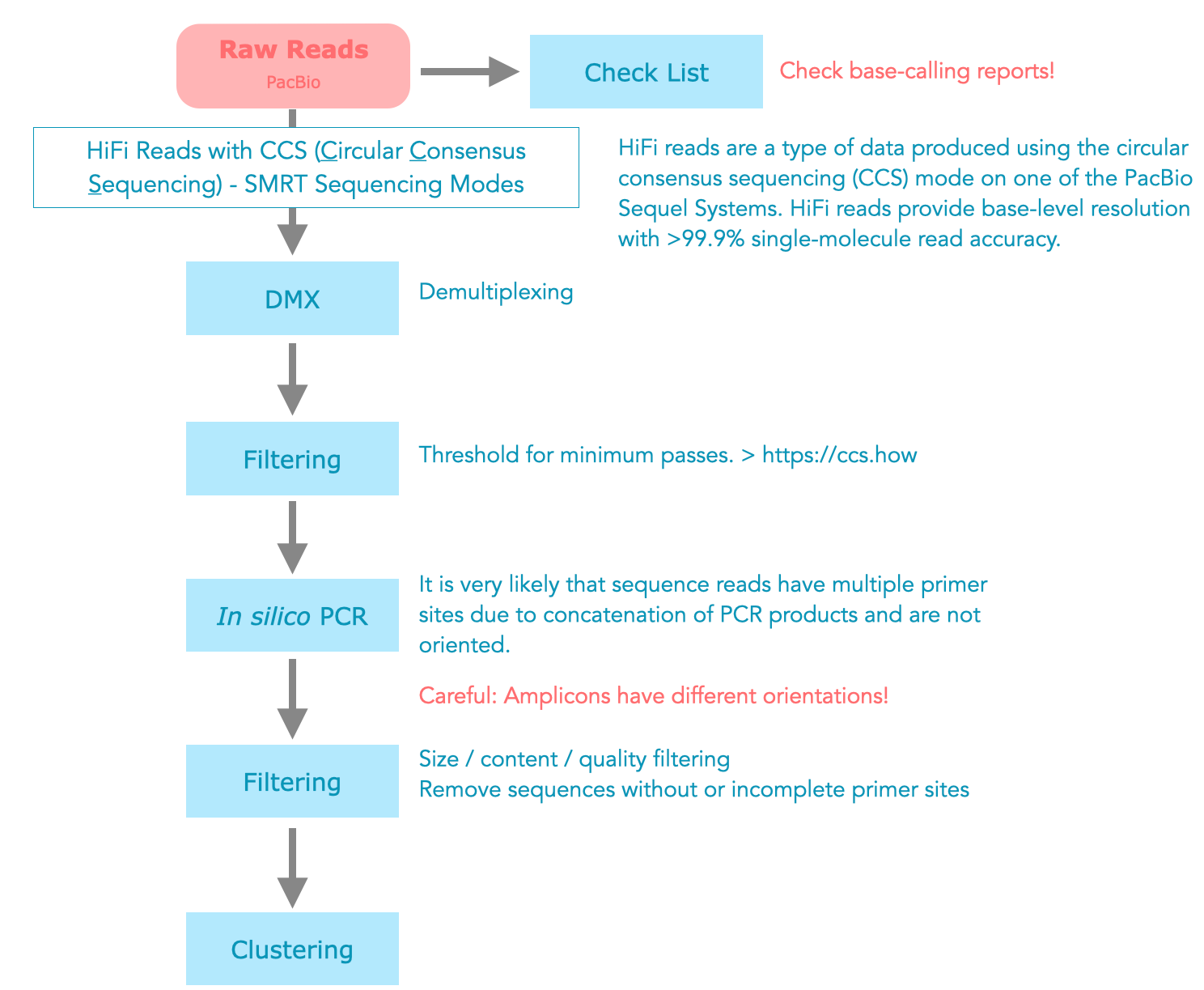
PacBio CCS Reads
More and more projects are using PacBio HiFi Reads (CCS). There are many different raw files per sample, but only one per sample is important for amplicon sequencing. In most cases this is a BAM file called CCS (Circular Consensus Sequencing) or HiFi (HiFi reads are produced using circular consensus sequencing (CCS) mode on PacBio long-read systems.).
<sample_name.**ccs**.bam>
PacBio Sequence Orientation
PacBio sequences are normally not oriented and need to be aligned. In addition, there are composite sequences with multiple primer sites. These must either be cleaned up or simply removed.
Element Bioscience (AVITI)
Another option has recently entered the market. EB's AVITI is a promising alternative to Illumina and we are currently gathering experience with it.
Workflow Overview
Illumina and PacBio data are not only different in read length. Therefore, the processing steps need to be adapted to the sequencing platforms. But even within the same technology, it is advisable to adapt the processing steps to the data structure.
Figure 1: Possible workflow scenarios for paired-end (Illumina) data.

Figure 2: Possible workflow for Pacbio CCS data.
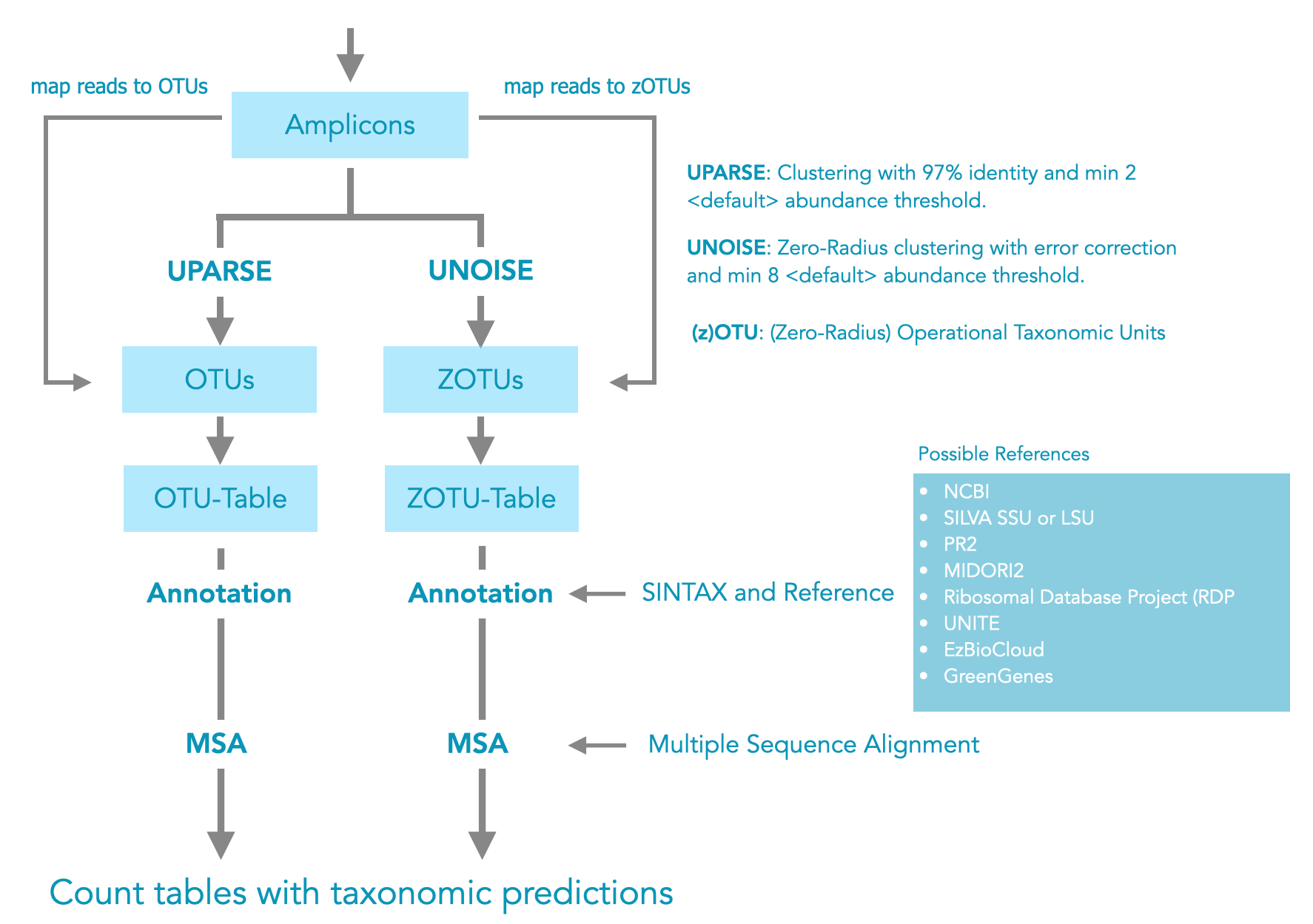
Once the data has been cleaned, clustering begins. Two different methods can be used to cluster the cleaned sequences (amplicons). On the one hand, it is possible to cluster sequences with a 3% radius (97% identity) (UPRASE: OTU). In this case, clusters with only one sequence (singletons) are usually removed. UPARSE does a very good job, but it should be noted that there are several equally good cluster solutions and not one best one. This means that repeating this step may slightly change the results. An alternative to 97% clustering is the zero-radius approach (UNOISE: zOTU). Here any sequence above a given abundance is used. Important in this approach is the error correction step before the reads are clustered. This approach is similar to the Amplicon Sequence Variant (ASV) method. In both clustering approaches, chimeras are removed by default.
Figure 3: Sequence clustering.
Preference
As I said, there are many good and not so good tools and approaches for processing AmpSeq data. I prefer USEARCH because it is a well-documented, very versatile and HPC-friendly amplication with excellent and long-standing support. True, USEARCH 64bit is not free and there are good alternatives (e.g. VSEARCH), but for sequencing and publishing we spend money without hesitation.
References
There are also many excellent references that can be used for taxonomic assignment. Some are maintained and available in different formats. The choice is yours, but be careful about mixing references.