
Learning Objectives
◇ Explain how amplicon sequencing (Amp-Seq) works and why controls are important.
◇ Recognize why different data preparation workflows are needed for different situations.
◇ Understand key steps in the data processing workflow and how they can be optimized.
◇ Identify potential problems with using "wrapper" tools that bundle multiple steps into one.
Amplicon Sequencing (Metabarcoding): Overview
Amplicon sequencing, also known as metabarcoding, is a targeted sequencing method used to study the composition of microbial communities. It involves amplifying and sequencing specific, highly variable regions of certain genes — regions that differ between species but are flanked by conserved sequences that are suitable for primer binding. Common marker genes include the 16S ribosomal RNA (rRNA) gene for bacteria and the internal transcribed spacer (ITS) region for fungi. These markers enable researchers to identify and compare species present in complex environmental samples.
Typical Amplicon Sequencing Workflow
1. Primer Design
Primers are short DNA sequences that target a specific genomic region (e.g., 16S or ITS). They must be specific enough to amplify the organisms of interest (e.g., bacteria or fungi) while avoiding off-target amplification. The chosen region should be highly variable to differentiate between taxa, but the length of the amplified region (amplicon) should remain relatively consistent to avoid sequencing bias.
2. PCR Amplification
The designed primers are used in a polymerase chain reaction (PCR) to amplify the target region. Successful amplification depends on optimal primer specificity, amplicon design, and well-tuned PCR conditions (e.g., annealing temperature, cycle number).
3. Library Preparation
The amplified DNA fragments are prepared for sequencing by attaching platform-specific adapter sequences and unique indices (barcodes) for each sample. This step enables multiplexing—sequencing many samples in a single run.
4. Sequencing
The prepared libraries are sequenced using high-throughput platforms such as Illumina (short-read, high-accuracy) or PacBio (long-read, useful for full-length amplicons). Millions of reads are generated in parallel, each representing a copy of the amplified region from the sample.
5. Data Processing (Preparation)
The raw sequencing reads are cleaned and processed using bioinformatics tools. This includes quality filtering, removing chimeras, and clustering or denoising the reads to generate count tables of operational taxonomic units (OTUs) or amplicon sequence variants (ASVs/zOTUs). These table show which sequences were found in which samples, and how often.
6. Data Analysis
Finally, the processed data is explored and interpreted. This involves identifying taxa, comparing the composition of communities between samples and testing ecological or experimental hypotheses.
Data Preparation
Once sequencing is complete, the raw data (usually in FASTQ format) must be processed before any meaningful analysis can be carried out. This step, known as data preparation or processing, involves cleaning, organising and structuring the sequencing data so that it can be used to identify the species or taxa present in each sample.
Unlike wet-lab protocols, the bioinformatics side of amplicon sequencing does not have a one-size-fits-all approach. Each dataset has its own particularities, such as different sequencing platforms, primers, read lengths, error profiles and sample types, so a fixed, universal workflow is rarely appropriate. Instead, we must carefully select and sometimes adapt the steps in our workflow to match the nature and quality of our data.
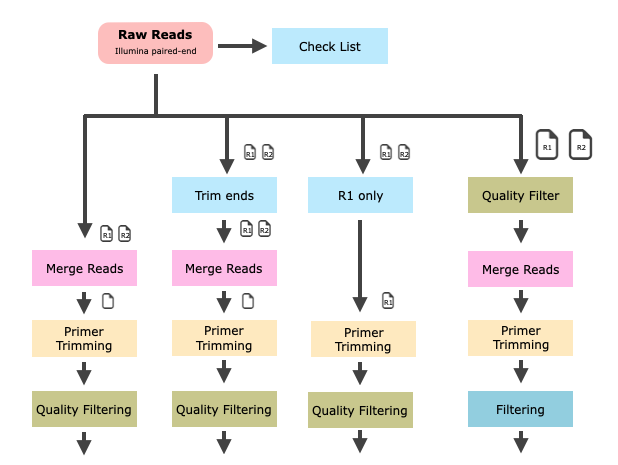
Why Not Just Use a Fixed Workflow?
It’s tempting to want a 'ready-made' pipeline that can perform all tasks with a single click. Although such tools (e.g. QIIME2, USEARCH pipelines and mothur) exist and can be useful, they may not always be the most effective or transparent option. If you rely solely on an automated black-box tool, you may miss important issues such as low-quality reads, poor primer trimming or unusual chimera patterns. Understanding each step of the workflow provides control and insight, which is particularly important when publishing results or troubleshooting.
Hands-on Exercise: Data Processing
In this practical session, you will take your first steps towards working with real amplicon sequencing data. The aim is to explore and inspect raw reads from an environmental sample, and begin processing them. You will work directly in the terminal, using basic Linux commands and bioinformatics tools.
If you're new to the command line, don't worry — we'll talk you through every step of the process.
Step 1: Get Ready
We’ll be working on a remote server that hosts the necessary tools and environment. Make sure you’re connected to the internet and have a terminal open.
## Log into the Server
ssh guest??@vserver.ethz.ch
# !!! Do not forget to replace `guest??` with the username assigned to you.
## Set Up Your Working Directory
mkdir -p ${HOME}/GDA/AmpSeq
cd ${HOME}/GDA/AmpSeq
## Download the Example Data
curl -O "https://www.gdc-docs.ethz.ch/GeneticDiversityAnalysis/GDA/data/SP05_R1.fastq.gz"
curl -O "https://www.gdc-docs.ethz.ch/GeneticDiversityAnalysis/GDA/data/SP05_R2.fastq.gz"
These are compressed FASTQ files containing paired-end sequencing reads from sample SP05.
## Verify the Downloads
md5sum SP05_R[12].fastq.gz
You should see:
2eeb689f8aadf31a5a47ada426830b6a SP05_R1.fastq.gz
c2961865c37078ede0903a1332bd36f0 SP05_R2.fastq.gz
Step 2: Explore the Raw Data
Before diving into processing, it’s helpful to understand what kind of data we’re working with. Try answering the following questions using basic command-line tools like zcat, head, and wc.
Here are some suggested commands and what they tell you:
What kind of data is it—single-end or paired-end?
You're working with paired-end data. You can confirm this by listing the two files:
ls SP05_R[12].fastq.gz
The _R1 and _R2 filenames indicate forward and reverse reads.
What is the read length distribution?
Check the sequence length using:
zcat SP05_R1.fastq.gz | head -n 2 | tail -n 1
This prints one read. Count the number of bases (or use wc -c to count characters):
zcat SP05_R1.fastq.gz | sed -n '2~4p' | head -n 5 | awk '{ print length($0) }'
This prints the lengths of the first few reads.
How many reads do we have?
Remeber, FASTQ files have 4 lines per read.
zcat SP05_R[12].fastq.gz | wc -l
Then divide the number by 4 to get the total read count. (Alternatively, we’ll do this programmatically later.)
What kind of sequences are these?
You can extract a few reads and copy them into NCBI BLAST to identify them:
zcat SP05_R1.fastq.gz | sed -n '2~4p' | head -n 3
How many unique instrument IDs to we have?
Check the instrument IDs in the headers of your FASTQ files to ensure that all reads originate from a single sequencing run. Mixed IDs may indicate technical variation or pooling, both of which can affect downstream analysis. If multiple IDs are present, consider accounting for them as potential batch effects.
curl -O "https://www.gdc-docs.ethz.ch/GeneticDiversityAnalysis/GDA/scripts/check_ids.sh"
bash check_ids.sh <(zcat SP05_R1.fastq.gz)
Challenge - Explore the Data Further
## QC report with: FastQC
fastqc SP05_R*.fastq.gz
# Have a look at the html reports.
## What kind of data?
# We have two files, R1 and R2. So, we expect paired-end reads.
# Let us explore the sequence header from the first and last record:
zcat SP05_R1.fastq.gz | head -n 1
zcat SP05_R2.fastq.gz | head -n 1
# header first R1 read: @M01072:31:000000000-A7V7A:1:1101:16534:1596 1:N:0:1
# |||||||||||||||||||||||||||||||||||||||||||| x||||||
# header first R2 read: @M01072:31:000000000-A7V7A:1:1101:16534:1596 2:N:0:1
zcat SP05_R1.fastq.gz | tail -n 4 | head -n 1
zcat SP05_R2.fastq.gz | tail -n 4 | head -n 1
# header last R1 read: @M01072:31:000000000-A7V7A:1:1103:27408:18721 1:N:0:1
# ||||||||||||||||||||||||||||||||||||||||||||| x||||||
# header last R2 read: @M01072:31:000000000-A7V7A:1:1103:27408:18721 2:N:0:1
## What is the read length?
zcat SP05_R1.fastq.gz | head -n 2 | grep "@" -v | awk '{print length($1)}'
# read length: 288nt
# => We are dealing with (Illumina MiSeq) PE-288 data.
## How many reads do we have?
zgrep -c "^+$" SP05_R[12].fastq.gz
# Both files have n(reads)=10'000
## What kind of sequences are we dealing with?
# get top 3 sequence in fasta format and run a NCBI nt blast.
zcat SP05_R1.fastq.gz | head -n 12 | grep "@M" -A 1 --no-group-separator | tr '@' '>'
# => Based on the results of the three blast searches,
# it can be assumed that this is a 16S rRNA amplicon of bacteria.
Step 3: Start Processing – Merging Paired Reads
Now that you have explored the raw data, it is time to start processing it. The first step is to merge the paired-end reads, combining each forward (R1) and reverse (R2) read to create a single, longer sequence.
Why do we do this?
- Amplicon sequencing often targets short regions (e.g. 250–500 bp), meaning the forward and reverse reads overlap in the middle.
- By aligning and merging the overlapping parts of each read pair, we can reconstruct the full-length amplicon.
- This improves data quality, as mismatches in the overlap region can highlight sequencing errors and merging can help to correct or remove them.
Tools for Merging Paired-End Reads
There are several tools available for merging paired-end reads—such as VSEARCH, USEARCH, and DADA2. Each has its own strengths and assumptions about how reads overlap.
In this exercise, we will use FLASh (Fast Length Adjustment of SHort reads):
- FLASh is a simple, fast, and effective tool designed specifically for merging short paired-end reads from next-generation sequencing.
- It works best when the overlapping region between forward and reverse reads is sufficiently long (typically ≥10–20 bp).
- It also provides useful (detailed) output metrics such as the number of successfully merged reads and the distribution of merged read lengths.
## Explore the application and parameters
flash --help
flash --help | less # page-by-page help - quit with [q]
flash --version
Make sure you understand the parameters that can affect merging quality:
- Overlap (min and max): How much overlap between reads is required to merge them.
- Mismatch tolerance: How many mismatches are allowed in the overlap.
- Read quality: Low-quality bases in the overlap region can lead to poor merging.
## Merge Reads - FLASh (default)
flash SP05_R1.fastq.gz SP05_R2.fastq.gz --threads 1
| FLASh (default) | Stats |
|---|---|
| Total reads | 10,000 |
| Combined reads | 4,583 |
| Uncombined reads | 5,417 |
| Percent combined | 45.8% |
The first default read-merging run with FLASh worked, but the merging success of ~46% seems low. There was also a warning message in the default run.
...
[FLASH] Read combination statistics:
[FLASH] Total pairs: 10000
[FLASH] Combined pairs: 4583
[FLASH] Uncombined pairs: 5417
[FLASH] Percent combined: 45.83% <<< !!!
[FLASH]
[FLASH] Writing histogram files.
[FLASH] WARNING: An unexpectedly high proportion of combined pairs (100.00%)
overlapped by more than 65 bp, the --max-overlap (-M) parameter. Consider
increasing this parameter. (As-is, FLASH is penalizing overlaps longer than
65 bp when considering them for possible combining!)
[FLASH]
[FLASH] FLASH v1.2.11 complete!
[FLASH] 0.146 seconds elapsed
[FLASH] Finished with 1 warning (see above)
Let's see if we can make the warning disappear and improve the merge rate:
- Flash wants us to adjust the
--max-overlap(default) parameter to > 65bp. We increase the maximum overlap length to read length. - We reduce the minimum required overlap from default 10 to 8.
- We increase the maximum mismatch density
## Merge Reads - using custom parameters (relaxed)
flash SP05_R1.fastq.gz SP05_R2.fastq.gz --threads 1 \
--min-overlap 8 \
--max-overlap 288 \
--max-mismatch-density 0.45
| Flash | Stats |
|---|---|
| Total reads | 10,000 |
| Combined reads | 5,397 |
| Uncombined reads | 4,603 |
| Percent combined | 54.0% |
A bit better but still not very good. We could further decrease the mismatch density and/or the minimum overlap.
## Merge Reads - super relaxed
flash SP05_R1.fastq.gz SP05_R2.fastq.gz --threads 1 \
--min-overlap 8 \
--max-overlap 288 \
--max-mismatch-density 0.95
| Flash | Stats |
|---|---|
| Total reads | 10,000 |
| Combined reads | 9,998 |
| Uncombined reads | 2 |
| Percent combined | 99.98% |
Great! More reads merged—problem solved, right?
Well… not quite.
By lowering the stringency of the merging parameters, we’ve made it easier for read pairs to merge, but this has likely come at the cost of some merging accuracy. Relaxing the overlap requirements means we risk joining reads that don’t actually belong together or contain more mismatches and errors, so more doesn’t always mean better.
Rather than relying on a brute-force approach to maximise numbers, we should take a closer look at the quality control (QC) report to evaluate the reliability of the merges.

As can be seen from the quality profile, read quality tends to decrease towards the end of the reads. This is a common issue in sequencing, especially with Illumina data, where the final bases are often less accurate.
But why does this matter?
Poor-quality bases at the ends of reads can interfere with merging by introducing mismatches, resulting in fewer or incorrect merges.
To address this issue, we will trim the low-quality ends of the reads prior to merging them. This reduces sequencing noise and increases the likelihood of accurate overlaps.
For this task, we will use `fastp, a fast and flexible quality-filtering tool with which you are already familiar from the quality filtering (QF) exercises. You will now see how it fits into a real amplicon sequencing workflow.
Please note: fastp is installed inside a Conda environment, so you’ll need to activate that environment before you can use the tool.
fastp depends on a number of other software libraries and tools in order to function correctly. Installing it system-wide could lead to version conflicts or missing dependencies, particularly on shared servers on which other bioinformatics tools are already installed.
Using Conda avoids these issues. Conda creates a self-contained environment in which all of fastP's dependencies are properly managed and isolated from the rest of the system. This makes the tool more stable and portable, and saves us from 'dependency hell'.
So, before running fastP, make sure you activate the correct environment.
## Load Conda Enironment
conda activate /usr/bin/condaenv/fastp
## Remember to use deactivate to switch between environments:
# conda deactivate
Once you have activated the environment, you will see that the prompt has changed, which confirms that the Conda environment has been loaded. You can then run fastp and any other tools installed in that environment.
fastp -v # version
fastp -h # help
To improve read quality and the success of merging, we’ll use fastp to trim the reads and remove those that are too short. This cleans up the low-quality bases at the ends and ensures that only informative reads are kept.
Run the following command:
## Trim Read and Remove short Reads
fastp --max_len1 240 \
-i SP05_R1.fastq.gz -I SP05_R2.fastq.gz \
-o SP05_R1_trim.fastq.gz -O SP05_R2_trim.fastq.gz
# => A detailed summary is printed on the screen.
--max_len1 240 ensures trimmed forward reads don't exceed 240 bp.
Let’s see if merging improves when we use the end-trimmed reads.
## Merge Reads - default
flash SP05_R1_trim.fastq.gz SP05_R2_trim.fastq.gz --threads 1 \
--max-overlap 240
| Flash (default) | Stats |
|---|---|
| Total reads | 9,884 |
| Combined reads | 9,860 |
| Uncombined reads | 24 |
| Percent combined | 99.76% |
Trimming the forward read (R1) significantly improved read merging, but take a closer look at the fastp report to see what actually changed.
Filtering result:
reads passed filter: 19768
reads failed due to low quality: 232
reads failed due to too many N: 0
reads failed due to too short: 0
reads with adapter trimmed: 7562
bases trimmed due to adapters: 55664
By default, fastp produces a lot of on-screen output. Can you find a way to redirect this output or generate a proper processing report instead?
Challenge - Can you redirect the output of `fastp` into a log file??
## Version A - simple minimal solution
fastp --max_len1 240 \
-i SP05_R1.fastq.gz -I SP05_R2.fastq.gz \
-o SP05_R1_trim.fastq.gz -O SP05_R2_trim.fastq.gz 2>&1 |\
tee log_fastp_230629.txt
# > tee copies standard input to standard output
## Version B - build-in solution
fastp --max_len1 240 \
--html log_fastp_230629.html \
--report_title Test \
-i SP05_R1.fastq.gz -I SP05_R2.fastq.gz \
-o SP05_R1_trim.fastq.gz -O SP05_R2_trim.fastq.gz
We have to be careful with the default settings in fastp! Not only are we trimming the reads, but we are also losing reads due to quality filtering (mean Q ≥ 15). By default, adapter trimming is also enabled. Let's disable both and focus on read end trimming only. We will also add a minimum read length filter.
## Trim Read and Remove short Reads
fastp --max_len1 240 \
--length_required 100 \
--disable_adapter_trimming \
--disable_quality_filtering \
-i SP05_R1.fastq.gz -I SP05_R2.fastq.gz \
-o SP05_R1_trim.fastq.gz -O SP05_R2_trim.fastq.gz
| fastp | Stats |
|---|---|
| Reads before | 10,000 |
| Reads after | 9,996 |
| Reads filtered | 0 |
We have a loss of 4 read pairs because they are too short after trimming. We could lower the default 15, but it probably wouldn't make sense.
fastp --help
# -l, --length_required > reads shorter than length_required will be discarded, default is 15. (int [=15])
Now, we will merge the trimmed reads with FLASh's default settings to see if trimming improves the merging process and allows us to compare the outcomes.
## Merge Trimmed Reads
flash SP05_R1_trim.fastq.gz SP05_R2_trim.fastq.gz
| FLASh default | Stats |
|---|---|
| Total reads | 9,996 |
| Combined reads | 9,667 |
| Uncombined reads | 329 |
| Percent combined | 96.7% |
Great! The merging process has improved significantly with FLASh’s default settings, and most read pairs are now successfully merged.
Now, let's take a closer look at the length distribution of the merged sequences using Emboss Infoseq, which will help us determine the sequence lengths.
infoseq -only -length out.extendedFrags.fastq | grep -v "Length" > amplicon_length.tmp
bash textHistogram.sh amplicon_length.tmp
The amplicons range in size from 211 nt to 468 nt, but the majority are around 288 nt.
211: #
...
286: #
287: #####
288: ##################################################
289: #######
290: #
...
468: #
We know that the error profiles of R1 and R2 reads are different. Since R2 is generally of lower quality and its quality deteriorates more quickly, different trimming strategies should be applied to each read. Trimming the poor-quality ends of the reverse reads (R2) might also improve the merging results further.
Challenge: Can you improve the merging even further by trimming the R2 reads as well?
## Trim Reads and Remove short Reads
fastp --max_len1 200 \
--max_len2 180 \
--length_required 100 \
--disable_adapter_trimming \
--disable_quality_filtering \
-i SP05_R1.fastq.gz -I SP05_R2.fastq.gz \
-o SP05_R1_trim.fastq.gz -O SP05_R2_trim.fastq.gz
## FastQC
#fastqc SP05_R1_trim.fastq.gz SP05_R2_trim.fastq.gz
## Merge Trimmed Reads
flash SP05_R1_trim.fastq.gz SP05_R2_trim.fastq.gz
# N(total) = 10,000
# N(filter)= 9,996 (fastp)
# N(merged)= 9,667 (previous)
# N(merged)= 9,836 (new)
Overlap |-| 46nt
R1 ----200--->
R2 <---180---
Amp |------288-------|
Finding the best parameters or data preparation strategy can be difficult. Running quality control (QC) before and after manipulating your reads can help you make informed decisions. Remember, it’s OK to lose some reads as long as you understand why.
With that in mind, we can now move on to the next step.
Primer Site Trimming
The primer site must be removed because it was added to the amplicon during PCR and is biologically irrelevant. However, before trimming, the output from FLASh must be renamed and the primer sequences obtained.
## Rename FLASh output
cp out.extendedFrags.fastq SP05_R12.fq
# I use the label R12 to indicate that
# pairs R1 and R2 have been merged.
## Verify the number of sequences
grep "^+$" -c SP05_R12.fq
# N: 9836 ✓
The primer sequences should be known from the targeted PCR:
Forward Primer: 5'-CAGCNGCCGCGGTAANAC-3' (l:18nt)
Reverse Primer: 5'-GGACTACNNGGGTNTCTAATC-3' (l:21nt)
Before trimming, we explore the primer sites using terminal commands.
Reminder: When searching for the reverse primer in merged reads, you must use its reverse complement primer sequence. Do you remember the terminal command for that?
Reverse-Complement Sequence
echo "GGACTACNNGGGTNTCTAATC" | rev | tr 'ACTG' 'TGAC'
When using grep to search nucleotide sequences, keep in mind that real FASTQ files usually don’t contain IUPAC ambiguity codes like 'N', 'Y', or 'R'. So, if your primer contains such characters, you'll need to replace them with appropriate regular expressions to match the actual bases in the sequences.
grep "CAGC[ACTG]GCCGCGGTAA[ACTG]AC" -c SP05_R12.fq
# N(PF): 8193 (83.3%)
grep "GATTAGA[ACTG]ACCC[ACTG][ACTG]GTAGTCC" -c SP05_R12.fq
# N(PF): 9516 (96.7%)
Challenge: Is there a way with grep to search for multiple primer sites?
grep itself searches for a single pattern at a time, but you can search for multiple patterns with the -E (extended regex) or -e options, like this:
sintax: grep -E "primer1|primer2"
grep -E "CAGC[ACTG]GCCGCGGTAA[ACTG]AC|GATTAGA[ACTG]ACCC[ACTG][ACTG]GTAGTCC" SP05_R12.fq
The grep approach is not ideal due to its limited tolerance for mismatches, but it provides a quick estimate. We see both primer sequences are present in the majority of amplicons, with the reverse primer found more frequently than the forward primer.
Now we use cutadapt to trim primer sites.
## Load Conda Environment
conda activate /usr/bin/condaenv/cutadapt
## Version
cutadapt --version
## Explore the many parameters of cutadapt
cutadapt --help
## Forward Primer Site Trimming
cutadapt -g CAGCNGCCGCGGTAANAC -e 0.1 -O 18 --trimmed-only -o SP05_R12_trimPF.fq SP05_R12.fq
grep "^+$" -c SP05_R12_trimPF.fq
# N(trimPF):9324 (94.8%)
The forward primer trimming works well because cutadapt accounts for possible sequencing errors in the primer region.
## Reverse Primer Site Trimming
cutadapt -a GATTAGANACCCNNGTAGTCC -e 0.1 -O 21 --trimmed-only -o SP05_R12_trimPF_trimPR.fq SP05_R12_trimPF.fq
grep "@M" -c SP05_R12_trimPF_trimPR.fq
# N(trimPR): 9304 (99.8%)
Almost 95% of amplicons contain the forward primer site and almost 100% contain the reverse primer site. Overall, this step results in a loss of 5.4% of amplicons.
This two-step trimming with cutadapt is useful for understanding what’s happening at each stage, but in practical pipelines, tools like USEARCH can streamline the process by detecting and trimming both primers at once using in-silico PCR.
Challenge: Can you figure out how to trim both primers in a single step using cutadapt?
cutadapt -g CAGCNGCCGCGGTAANAC \
-a GATTAGANACCCNNGTAGTCC \
-e 0.1 -O 15 \
--discard-untrimmed \
-o SP05_R12_trimmed.fq SP05_R12.fq
Clustering
We are using USEARCH for the clustering. The sequence clusters are called operational taxonomic units (OTU). An OTU is an operational definition used to classify groups of related sequences. These are not species and OTUs do not exist in nature. They are ad-hoc clusters made for convenience.
We could either use the classical clustering approach with a 97% similarity threshold to obtain Operational Taxonomic Units (OTUs). We typically remove singletons (unique amplicons) and apply a minimum abundance threshold of 2. This clustering approach has been criticised because it often does not produce a single best solution and because closely related species can be clustered together. Alternatively, we could assume that each unique sequence is a potential 'species', but apply a higher abundance threshold to account for sequencing errors. The resulting OTUs are called Amplicon Sequence Variants (ASVs) or Zero Radius OTUs (zOTUs) if you are using UNOISE.
## De-replicate Amplicons
usearch -fastx_uniques SP05_R12_trimPF_trimPR.fq \
-fastaout SP05_Uniques.fa \
-sizeout
# Have a closer look at the stats:
# 9304 seqs, 4644 uniques, 3330 singletons (71.7%)
# Min size 1, median 1, max 221, avg 2.00
## Create 97% similarity clusters > OTUs
usearch -cluster_otus SP05_Uniques.fa \
-zotus SP05_OTU.fa \
-minsize 2
## Create (error-corrected) zero-radius OTUs (similar to amplicon sequence veriants (ASVs))
usearch -unoise3 SP05_Uniques.fa \
-zotus SP05_zOTU.fa \
-minsize 3
grep "^>" -c SP05_zOTU.fa
# => 636 zOTUs
To obtain the counting tables, we now need to map the merged and primer-trimmed amplicons to the clusters (e.g. zOTUs).
## Make count tables
usearch -otutab SP05_R12_trimPF_trimPR.fa -otus SP05_zOTU.fa -otutabout SP05_zOTU.tab
# 7019 / 9836 mapped to zOTUs (71.4%)
Taxonomic Assignments
Now we have a count table with (z)OTUs as rows and sample(s) as columns. In a next step we try find taxonomic associations with known species.
## Reference
REF="/usr/data/rdp_16s_v18.fa"
## Data Format Reference
head -n 4 ${REF}
## Size Reference
grep ">" -c ${REF}
# N = 10,049
## Taxonomic Prediction
usearch -sintax SP05_zOTU.fa \
-db ${REF} \
-strand both \
-sintax_cutoff 0.85 \
-tabbedout SP05_zOTU_RDP.tax
## Example
grep "Zotu1" -w SP05_zOTU_RDP.tax
# => d:Bacteria,p:Acidobacteria,c:Acidobacteria_Gp4,g:Gp4
The most common OTU (Zotu1) could represent an Acidobacteria subgroup Gp4. Let us see if we can confirm the annotation using NCBI BLAST. By default, NCBI BLAST is using the entire nucleotide collection (nr/nt) producing rather unspecific hits. I recommend to use the reference RNA sequences database instead.
grep "Zotu1" -w -A 4 SP05_zOTU.fa
>Zotu1
GGGGGGAGCAAGCGTTGTTCGGATTTACTGGGCGTAAAGGGCGCGTAGGCGGTCAGCACAAGTCAGTTGTGAAATCTCCG
GGCTTAACTCGGAAAGGTCAACTGATACTGTGCGACTAGAGTGCAGAAGGGGCAACTGGAATTCTCGGTGTAGCGGTGAA
ATGCGTAGATATCGAGAGGAACACCTGCGGCGAAGGCGGGTTGCTGGGCTGACACTGACGCTGAGGCGCGAAAGCCAGGG
GAGCGAACGG
We got good but not perfect hits. The top 5 blast hits are equally good:
| Accession | Species | Coverage | Identity | Taxa |
|---|---|---|---|---|
| NR_151987.1 | Brevitalea aridisoli | 100% | 93.6% | Bacteria;Acidobacteria;Blastocatellia;Blastocatellales;Pyrinomonadaceae;Brevitalea |
| NR_151988.1 | Brevitalea deliciosa | 100% | 93.6% | Bacteria;Acidobacteria;Blastocatellia;Blastocatellales;Pyrinomonadaceae;Brevitalea |
| NR_151986.1 | Arenimicrobium luteum | 100% | 93.6% | Bacteria;Acidobacteria;Blastocatellia;Blastocatellales;Pyrinomonadaceae;Arenimicrobium |
| NR_146026.1 | Tellurimicrobium multivorans | 98% | 91.5% | Bacteria;Acidobacteria;Blastocatellia;Blastocatellales;Blastocatellaceae;Tellurimicrobium |
| NR_146021.1 | Stenotrophobacter namibiensis | 98% | 91.5% | Bacteria;Acidobacteria;Blastocatellia;Blastocatellales;Blastocatellaceae;Stenotrophobacter |
The top 5 blast results would agree on the following annotation for zOTU1:
d:Bacteria,p:Acidobacteria,c:Blastocatellia,o:Blastocatellale
It seems we improved the taxonomic prediction if we can trust the NCBI references.
As an alternative to a Blast-Search we could use SILVA's SINA aligner. A classification service specific for SSU sequence fragments.
# OTU1 d:Bacteria;p:Acidobacteriota;c:Blastocatellia;o:Pyrinomonadales;f:Pyrinomonadaceae;RB41
The results agree mostly with the blast results with the exception for the order. It seems that the order label is inconsistent between references.
Taxonomic Assignments
The taxonomic classification of OTUs is a difficult and error-prone process. There are many reasons for this. First, there is no perfect reference, and those that are available are incomplete and contain errors. The amplicon may be too short or not diverse enough to provide sufficient taxonomic resolution. Taxonomic assignments are always predictions and should never be blindly trusted.
Data Analysis
Challenges
For the exercises you need to download and unzip the R script, the data, and some "homebrew" functions. All files are combined in one zipped file. Below are all the steps in R you need to download, unzip, and open the R script. Have fun!
## You need this R library to use wget
library("HelpersMG")
## Working directory
getwd()
## Create a new working folder (if needed)
# dir.create("~/GDA")
# dir.create("~/GDA/AmpSeq")
## Change to new working directory
setwd("~/GDA/AmpSeq")
## Confirm new working directory
getwd()
## Download data (R data image file)
data.url <- "https://www.gdc-docs.ethz.ch/GeneticDiversityAnalysis/GDA/data/AmpSeqExample.zip"
utils::download.file(data.url, destfile = "AmpSeqExample.zip")
unzip("AmpSeqExample.zip")
## Have a look at the folder content
list.files()
# These are the files you should see in your working directory:
# (1) AmpSeq.zip: your original data file
# (2) AmpSeqExercises.R: R script
# (3) Chaillou2015.Rdata: input file
# (4) graphical_methods.R: file with various functions
## Remove File and Directory
file.remove("AmpSeqExample.zip")
## Load R script
file.edit("AmpSeqExercises.R")
AmpSeq Data-Prep Workflows and more ...
- UPARSE & UNOISE
- Quantitative Insights Into Microbial Ecology - QIIME
- mothur project
- DADA2 pipeline
- OBITools
- nextflow - nf-core/ampliseq
Data Analysis
- Phyloseq - analysis of microbiological communities
- microbiome R package
- ampvis2
- vegan: an R package for community ecologists
- mixOmics