
This page contains a collection of small but important topics that don’t fit neatly into other sections. Consider it a reference page for quick access to key information. If you think anything is missing, feel free to let us know!
File Extensions
File extensions (sufix) are critical to the proper functioning of both users and systems, enabling efficient file management, ensuring security, maintaining compatibility and facilitating automated processes. It is therefore important to specify the file type and to do so correctly.
# Text File:
echo "Test" > file.txt
# Temporary Test File
echo "Temp-Test" > file.tmp
# Sequence File:
echo ">Seq01\nATGCCGTCACCGT" > file.fa
Naming
You can name your files and folders anything you like. However, following naming conventions can help maintain a tidy, efficient, and easily navigable digital environment, reducing the risk of misplaced files and making it easier for everyone to find and manage documents.
General Rules & Best Practices
- Meaningful but short and descriptive
- Use consistent naming conventions
- Avoid special characters
- Use leading zeros for sorting
- Indicate versions and revisions
- Consider case sensitivity
- Avoid redundancy
Here are some examples of the do's and don'ts of naming.
Balance length and clarity
Bad: Genetic Diversity Analysis - Rooling Dice Exercise.txt
Better: GDA_RoolingDice_BobaFett_V01.R
Naming convention and numerical order
Bad: water_sample_river_12.fa
Better: RW-012.fa
No space
# Does not work
echo "Test" > My First File
# Works but it's not a good idea
echo "Test" > "My First File"
# Also works but it's still not a good idea
echo "Test" > My\ First\ Test
# What you should use instead
echo "Test" > MyFirstTest.txt # camel case
echo "Test" > my_first_test.txt # snake case
The same applies to folders.
# Bad idea
mkdir "Material and Methods"
# Does not work:
cd Materials and Methods
# Works but it is not optimal:
cd Materials\ and\ Methods
cd "Material and Methods"
# Prefer underscores or hyphens over spaces:
mkdir Material_and_Methods
cd Materials_and_Methods
You are up. Find better solutions for the following file names:
- Spacing and case sensitivity
my new file
a) ⇒ mynewfile.txt (simple but difficult to read)
b) ⇒ MyNewFile.txt (simple and easier to read > camel case)
c) ⇒ My_New_File.txt (clear but needs more space > snake case)
Note #2: Be aware of case sensitivity. ```
- Numbers
Sample1 (ph5) temperatur high.fasta
a) ⇒ Sample01_pH5_High.fa
b) ⇒ S01_ph5_hi.fa
file1.txt file1.txt
file2.txt -sort-> file10.txt
file10.txt file2.txt
file01.txt file01.txt
file02.txt -sort-> file02.txt
file10.txt file10.txt
# Problem with sorting file names numerically
touch f1.txt f02.txt f123.txt f1234.txt
ls -1
# f02.txt
# f1.txt
# f123.txt
# f1234.txt
# Loop to fix file name
for file in f*.txt
do
# Adjust sed according to you file name
number=`echo ${file%.*} | sed 's/f//g'`
suffix=${file##*.}
# Adjust length (4) accordingly
new_name=`printf f%04d.%s ${number} ${suffix}`
mv $file $new_name
done
#f0001.txt
#f0002.txt
#f0123.txt
#f1234.txt
- Special characters
GDA Final Report: Irène Müller.md
⇒ GDA24_FinalReport_Irene_Mueller.md
- What about dots?
No need to use . (dot) in a filename execpt dots improves readability of filenames.
zip file.zip primer*.fq
✕ file.zip (file type is lost)
⇒ primer.fa.zip
cp setting.txt settings.txt.backup
- Keep it short
# Messy Looking Sample List:
sample1_rive_SG.fa
sample02_lake_SG.fasta
s3_water-lake_Zurich.fa
sample4_water_lake_ZH.fasta
sam5_water-river_TG.fa
# Nice Looking Sample List:
S01_R_SG.fa
S02_L_SG.fa
S03_L_ZH.fa
S04_L_ZH.fa
S05_R_TG.fa
File Formats
FASTA
The FASTA format is a text-based format for representing one or more nucleotide or protein sequences. The first line (header) of a FASTA record starts with a greater-than symbol, followed by a unique description of the sequences (e.g. accession number). The header is followed by the actual sequence as a string, spread over one or more lines.
Multiple-Line (Wrapped) Sequence Fasta File:
>Sequence_001
ATGGGCGTCACGTCCACGTTCACCGTGTTA
TGGAATGCGTCACGTCAACTGGGGT
>Sequence_002
ATGGCCGTCACGTCCACGTTCACCGTGTTATGGAATGCGACACGTCAACTGGGGT
>Sequence_003
ATGGCCGTCACGTCCACGTT
Single-Line Sequence Fasta File:
>Sequence_001
ATGGGCGTCACGTCCACGTTCACCGTGTTATGGAATGCGTCACGTCAACTGGGGT
>Sequence_002
ATGGCCGTCACGTCCACGTTCACCGTGTTATGGAATGCGACACGTCAACTGGGGT
>Sequence_003
ATGGCCGTCACGTCCACGTT
To count the number of sequences in a Fasta file, it is best to count all lines beginning with a > sign.
grep -c ">" file.fa
zgrep -c ">" file.fa.gz
0.5 * n(lines) ≠ n(sequences)
The number of lines divided by two is not necessarily the number of sequences in a multi-sequence fasta file.
FASTQ
A FASTQ file is a text-based format for storing both a biological sequence (usually nucleotide sequences) and its associated quality scores.
Each entry (read/sequence) in a FASTQ file consists of 4 lines:
1: @M01072:41:000000000-A942B:1:1101:11853:2457 1:N:0:1
2: GTGCCAGCAGCCGCGGTAATACGTAGGTGGCAAGCGTTATCCGGATTTATTGTGCGTAAAGGGAACGC...
3: +
4: >>1>>11>11>>1EC?E?CFBFAGFC0GB/CG1EACFE/BFE///AEG1DF122A/B///21//0BEG...
1: Header Line - Unique sequence identifier.
2: Sequence Line - The nucleotide sequence (the base calls; A, C, T, G and N).
3: Plus Line - A separator, which is simply a plus (+) sign.
4: Quality Line -The base call quality scores. These are Phred +33 encoded,
using ASCII characters to represent the numerical quality scores.
To count the number of sequences in a Fastq file, you have to be careful. Counting the line with a @ sign may not work because the quality line may also contain pund signs. We could be a bit more specific in our search and only include cases where the line starts with a delimiter, and we could also include part of the header like grep -c "^@M01072" file.fq. However, this would only work if all the sequences had the same instrument name (M01072). But we could take advantage of the fact that we always have four lines per record.
## Number of lines / 4
echo $(($(wc -l < file.fq) / 4))
## Lines starting and ending with + (Not pulletproof)
zgrep -c "^+$" file.fq.gz
0.25 * n(lines) = n(sequences)
The number of lines divided by four equals the number of sequences in a multi-sequence fastq file.
FASTG
Fastg is a file format for the faithful representation of genome assemblies. The G stands for graph. Genomes are represented as linear sequences stored in FASTA files. This makes sense as long as we know the genome. However, many genomes contain polymorphisms that cannot be described in a linear sequence, and almost all assemblies contain errors and problematic regions. The FASTG format aims to solve this problem with a flexible, graph-based approach to encoding all variability in the sequence. Bandage is a program for visualising de novo assembly graphs using fastg input files. Assembly graphs in the FASTG format can be converted to linear FASTA sequences.
Example for a fastg file (Metagenome assembler: Megahit)
>NODE_12427_length_4458_cov_84.0000_ID_24853':NODE_164443_length_1445_cov_44.3323_ID_328885',NODE_515499_length_44875_cov_57.9870_ID_1030997';
CGCTTGCTCGATTTGACCCCATAAGCGGAATCACCACCCAATAGCATGTTGCCGCCAGAAGCAGAACCCCGTATTTCTTCTAAAGACTCTCCTAATTGTTCCAGAGCAACTAGAAAGAGATTCTTTATTTTCTCGTCTTTATTGTAGCTA...
>NODE_515499_length_44875_cov_57.9870_ID_1030997:NODE_12427_length_4458_cov_84.0000_ID_24853;
CTTTGCTTTTGTAATGGTGCCTTGCAATAAATTCTACAAGAGCTAAAAAAGAGTCTCATCATTCACGACTAATCTTCTATATCGTAGATGCTTGT...
>NODE_515499_length_44875_cov_57.9870_ID_1030997';
TCAAAATATGGAACGCCTACTTTAACTTTCTTTGTGATGTCTTCTTCAGTGT...
FAST5 / POD5
A FAST5 file is a hierarchical data format (HDF5) used to store raw data and metadata from nanopore sequencing technologies such as those produced by Oxford Nanopore Technologies.
POD5 is a file format developed by Oxford Nanopore to store nanopore data in an accessible way, replacing the legacy .fast5 format. This output also reads and writes data faster, uses less computing power and has a smaller raw data file size than .fast5.
Raw current signal data are generated on an ONT sequencing device are usally base-called into fastq sequence reads.
SAM / BAM
SAM (Sequence Alignment/Map) and BAM (Binary Alignment/Map) are file formats to store DNA sequence alignment data.
SAM files are plain text files that store sequence alignment information. They contain a header section and an alignment section. The header provides information about the reference genome used, sequence dictionary, and other metadata. The alignment section contains records for each aligned read, including details such as the read ID, reference sequence name, alignment position, quality scores, flags indicating alignment properties, and more.
@HD VN:1.6 SO:coordinate
@SQ SN:chr1 LN:248956422
@RG ID:sample1 LB:lib1 SM:sample1 PL:illumina
@PG ID:bwa PN:bwa VN:0.7.17-r1188 CL:bwa mem -t 4 ref.fa read1.fq read2.fq
read1 99 chr1 1000 30 10M1D20M = 2000 0 AGCTTAGCTAGCTAGCTAGCTAGCTAGCTA IIIIIIIIIIIIIIIIIIIIIIIIIIIIIII NM:i:1 MD:Z:10^C20 AS:i:45 XS:i:40
read2 147 chr1 2000 30 30M = 1000 0 CAGCTAGCTAGCTAGCTAGCTAGCTAGCTA IIIIIIIIIIIIIIIIIIIIIIIIIIIIIII NM:i:0 MD:Z:30 AS:i:45 XS:i:50
-
The first line starting with "@HD" provides header information, including the version (VN) and the sort order (SO).
-
The following lines starting with "@SQ" define the reference sequences (chromosomes), specifying their names (SN) and lengths (LN).
-
The "@RG" line represents read group information, including an ID, library name (LB), sample name (SM), and platform information (PL).
-
The "@PG" line describes the program used for alignment, including its ID, name (PN), version (VN), and command-line (CL) parameters.
-
The subsequent lines represent alignment records for individual reads and each line consists of several fields separated by tabs. The fields provide information such as read ID, alignment flags (99 or 147 in this case), reference sequence name (chr1), alignment position (1000 or 2000), mapping quality (30), CIGAR string representing the alignment (10M1D20M or 30M), mate information (= symbol and mate's position), and more. The last fields contain tags with additional information, such as the edit distance (NM), mismatch positions (MD), alignment score (AS), and supplementary alignment score (XS)
BAM files, on the other hand, are binary versions of SAM files. They are compressed and indexed, which makes them more efficient in terms of storage and retrieval speed compared to SAM files. BAM files are widely used in practice due to their smaller file size and faster processing times.
VCF
A VCF (Variant Call Format) file is a standard file format for storing genetic variations, such as single nucleotide polymorphisms (SNPs), insertions, deletions, and structural variants. Each record in a VCF file includes various fields that provide details about the genetic variation, such as the chromosome location, reference allele (the allele present in the reference genome), alternative allele(s) (the observed variant allele(s)), quality scores, genetic annotations, and genotype information for individuals or samples.
##fileformat=VCFv4.3
##fileDate=2023-06-28
##source=GenomeAnalyzerV2.0
##reference=GRCh38
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT SAMPLE1 SAMPLE2 SAMPLE3
1 100 rs123 A T 100 PASS . GT 0/1 1/1 0/0
1 200 rs456 G C 50 PASS . GT 1/1 0/0 1/1
1 300 rs789 C T 80 PASS . GT 1/1 1/1 0/1
-
The lines starting with "##" are metadata lines providing information about the file format, date, source, and reference genome used.
-
The line starting with "##FORMAT" describes the format of the genotype field.
-
The line starting with "#CHROM" contains the column headers, specifying the fields present in the VCF file. In this case, it includes chromosome (CHROM), position (POS), variant ID (ID), reference allele (REF), alternative allele (ALT), quality score (QUAL), filter status (FILTER), additional information (INFO), format of the genotype field (FORMAT), and the samples (SAMPLE1, SAMPLE2, SAMPLE3).
-
The subsequent lines represent individual variant records, each describing a specific genetic variation.
For example, the first variant is located on chromosome 1 at position 100. The reference allele is 'A', and the alternative allele is 'T'. The quality score is 100, and it has passed the filtering criteria. The format of the genotype field is GT, and the genotype for the three samples is 0/1, 1/1, and 0/0, respectively.
This is a simplified example, and VCF files can contain additional fields and annotations depending on the specific analysis and requirements.
Encoding
A code is a system of rules for translating information. For example, the genetic code is a sequence of codons that correspond to a sequence of amino acids. In other words, a gene can code for a protein. Character encoding in textual data is used to represent a repertoire of characters. A common example of a character encoding system is Morse code. We use Morse code to encode text characters as standardised sequences of dots and dashes.
--. -.. -.-. (➞ GDC)
Another example is ASCII or Unicode (UTF), which are character encoding standards for electronic communications. Both ASCII and Unicode (UTF) are essential for electronic communications, with ASCII being suitable for basic text and control codes, and Unicode providing extensive support for global text representation.
| binary code | ASCII code | Letter |
|---|---|---|
| 01000111 | (64 + 4 + 2 + 1 =) 071 | G |
| 01000100 | (64 + 4 =) 068 | D |
| 01000011 | (64 + 2 + 1 =) 067 | C |
Plain text files need the corresponding encoding to decode the message correctly. You can use the terminal command file to determine the file type:
file -bi file.txt
Note: Unicode is a superset of ASCII (7 bits). The main difference is in the way they encode the character and the number of bits used.
Especially the ambiguity of control characters are notorious for causing troubles. A good example is the newline (end of line (EOL), line breaks) encoding.
| OS | EOL | |
|---|---|---|
| Linux | LF | /n |
| macOS | LF | /n |
| MS Windows | CRLF | /r/n |
A friendly tip
Having trouble importing files? Double-check the file encoding! Different systems and programs use various encodings, such as UTF-8 or ISO-8859-1, and mismatches can cause errors. Ensure that both the file and the program you're using are set to the same encoding to avoid issues. If you're unsure, try opening the file with a text editor to confirm the encoding or check your program's import settings.
K-mers
In bioinformatics, K-mers are substrings of length k of biological sequences (e.g. nucleotides of DNA sequences). The abundance of a set of k-mers in the genome of a species, in a genomic region, or in a class of sequences can be used as a "signature" of the underlying sequence. Comparison of these abundances is computationally simpler than sequence alignment and is an important method in non-alignment sequence analysis. It can also be used as the first stage of analysis before alignment.
Example: k-mers for DNA string GTC
| k | k-mer(s) |
|---|---|
| 1 | G, T, C |
| 2 | GT, TC |
| 3 | GTC |
Compress Sequence Files
Sequencing centres provide compressed sequence files for good reasons. Compressed files are smaller and more secure to download. Keep the data compressed, as many applications can handle compressed files directly. If you need to decompress data, consider using scratch space if available. In general, compress both fastq and fasta sequence files (i) to save disk space and (ii) for safer data transfer.
Linux has several commands for compressing and decompressing files. We will take a closer look at gzip and tar.
## Working Directory
mkdir -p ${HOME}/MDA/qc
cd ${HOME}/MDA/qc
## Toy Dataset
# Use either `wget` or `curl` to download the paired-end sequences.
#wget "https://www.gdc-docs.ethz.ch/GeneticDiversityAnalysis/GDA/data/C-1_R1.fastq.gz"
#wget "https://www.gdc-docs.ethz.ch/GeneticDiversityAnalysis/GDA/data/C-1_R2.fastq.gz"
curl -O "https://www.gdc-docs.ethz.ch/GeneticDiversityAnalysis/GDA/data/C-1_R1.fastq.gz"
curl -O "https://www.gdc-docs.ethz.ch/GeneticDiversityAnalysis/GDA/data/C-1_R2.fastq.gz"
wget or curl?
Both wget and curl are command-line tools used for transferring data over various network protocols. While they have some similarities, they also have distinct differences. wget is focused on downloading files from the web and provides more detailed progress information, while curl is a versatile tool that supports various protocols, offers more flexibility, and can be used for more complex tasks beyond file downloading.
There are terminal commands able to handle commpressed files. You used cat to print the content of text files either to the screen or a file. The command zcat is doing the same but works for compressed file.
## Create a test file - subset
zcat C-1_R1.fastq.gz | head -n 40 > subset_C-1_R1.fq
# zsh: zcat < C-1_R1.fastq.gz | head -n 40 > subset_C-1_R1.fq
# zsh: gzcat C-1_R1.fastq.gz | head -n 40 > subset_C-1_R1.fq
head -n 4 subset_C-1_R1.fq
wc -l subset_C-1_R1.fq
zcat C-1_R2.fastq.gz | head -n 40 > subset_C-1_R2.fq
# zsh: zcat < C-1_R2.fastq.gz | head -n 40 > subset_C-1_R2.fq
# zsh: gzcat C-1_R2.fastq.gz | head -n 40 > subset_C-1_R2.fq
tail -n 4 subset_C-1_R2.fq
wc -l subset_C-1_R2.fq
Shell differences
The command zcat works differntly under bash shell and zsh. Use gzcat or use < with zcat.
zcat C-1_R1.fastq.gz
zcat: cannot stat: C-1_R1.fastq.gz (C-1_R1.fastq.gz.Z): No such file or directory
zcat works different on your Mac! Try the following instead:
zcat < C-1_R1.fastq.gz | head -n 40 > subset_C-1_R1.fq
Random sampling
The head -n 40 command is not a perfect way to get a subset because it takes only the top sequences and not a random subset! A better alternative provides e.g., seqtk:
seqtk sample -s1505 C-1_R1.fastq.gz 10 > random_subset_C-1_R1.fq
seqtk sample -s1505 C-1_R2.fastq.gz 10 > random_subset_C-1_R2.fq
Note: Use the seed option to keep the paired reads in sync.
GNU Zip
## ----------------------
## GNU zip - compress
## ----------------------
## Zip file
gzip subset_C-1_R*.fq
## Compression info
file subset_C-1_R*.fq.gz
## Compression level
gzip -l subset_C-1_R*.fq.gz
## ----------------------
## GNU zip - un-compress
## ----------------------
## Un-zip file
gunzip subset_C-1_R*.fq.gz
## ----------------------
## GNU zip - Compression level
## ----------------------
# You can specify the compression levels from 1 (fast) - 9 (best).
# The default level is 6.
## Speed-Test
# Test data
cp C-1_R1.fastq.gz test.fq.gz
gunzip test.fq.gz
# Level 1 (fast)
time gzip -1 test.fq
# t = 1.781s to zip at level 1 with 65.2% compression rate
time gunzip test.fq.gz
# t = 1.099s
# Level 6 (default)
time gzip -6 test.fq
# t = 8.459s to zip at level 6 with 74.4% compression rate
time gunzip test.fq.gz
# t = 0.864s
# Level 9 (best compression)
time gzip -9 test.fq
# t = 32.012s to zip at level 9 with 75.6% compression rate
time gunzip test.fq.gz
# t = 0.982s
# Clean what we do not need any longer
rm test.fq
# ⇒ Default works best for our example in terms of speed and compression.
## ----------------------
## GNU zip - Help
## ----------------------
man gzip
gzip --help
TAR Archive
## ----------------------------
## TAR - uncompressed archive
## ----------------------------
## tar (create archive)
tar cfv subset_C-1_R1R2.fq.tar subset_C-1_R1.fq subset_C-1_R2.fq
## Change in size
du -sch subset_C-1_R1R2.fq.tar subset_C-1_R1.fq subset_C-1_R2.fq
# The files are archvied together but not compressed:
# 20K subset_C-1_R1R2.fq.tar
# 8.0K subset_C-1_R1.fq
# 8.0K subset_C-1_R2.fq
## un-tar (extract)
tar xfv subset_C-1_R1R2.fq.tar
## ----------------------------
## TAR - compressed archive
## ----------------------------
## tar with compression (option z)
tar cfvz subset_C-1_R1R2.fq.tar.gz subset_C-1_R1.fq subset_C-1_R2.fq
du -sch subset_C-1_R1R2.fq.tar.gz subset_C-1_R1.fq subset_C-1_R2.fq
# The files are archived and compressed
# 8.0K subset_C-1_R1R2.fq.tar.gz
# 8.0K subset_C-1_R1.fq
# 8.0K subset_C-1_R2.fq
## un-tar and de-compress
tar xfvz subset_C-1_R1R2.fq.tar.gz
## ----------------------------
## TAR - Help
## ----------------------------
man tar
tar --help
info tar
Exploring Without Decompression
You don’t always need to fully decompress a file to check what’s inside. Here are some simple terminal commands to help you quickly peek into compressed files without extracting them.
The zcat command allows you to view the contents of a gzipped file without extracting it:
zcat C-1_R1.fastq.gz | head -n 4
This will display the first four lines of the file. You can replace 4 with any number to view more or fewer lines.
(2) Another option is using gzip with the -c and -d flags:
gzip -cd C-1_R1.fastq.gz | head -n 4
-c: Output to the terminal, keeping the original file unchanged.-d: Decompress the file (but only for viewing, without saving an uncompressed version).
(3) If you’re dealing with .tar.gz archives, you can list the contents without unpacking the files using:
tar -tf C-1_R1.fastq.tar.gz
-t: List the contents of the archive.-f: Specify the archive file to read from.-v: (Optional) Show details in a more verbose output.
(4) For .zip files, the zipinfo command provides detailed information about the contents, such as file names, sizes, and modification dates:
zipinfo file.zip
This command gives you an overview of everything inside the .zip file, including file permissions, compressed size, and more.
Watch Disk Space
Be careful when unpacking the file(s). Make sure that there is enough disk space available (e.g. df -h). Consider using disk space that is not backed up (e.g. scratch). Zip large files and tar small files to keep your project folder tidy and manageable.
File Integrity
Every time you transfer data, there is a chance that the file will be corrupted. To check the integrity of a file, we can use md5sum. This command calculates a message digest fingerprint (checksum) for a file.
md5sum C-1_R[12].fastq.gz
# 4a2e8876742302fad5bd24cba76c3cc6 C-1_R1.fastq.gz
# e0ce4ae266e8b2cedbc9580a3025712a C-1_R2.fastq.gz
Not working for you?
You might have to use a differnt function.
Try md5 instead: md5 C-1_R[12].fastq.gz
Changing the filename of an archived file does not change the checksum. The file name is not important:
cp C-1_R1.fastq.gz test.fq.gz
md5sum C-1_R1.fastq.gz test.fq.gz
# 4a2e8876742302fad5bd24cba76c3cc6 C-1_R1.fastq.gz
# 4a2e8876742302fad5bd24cba76c3cc6 test.fq.gz
rm test.fq.gz
Changing the file content, changes the key:
gunzip -c C-1_R1.fastq.gz > test.fq
gzip test.fq
md5sum C-1_R1.fastq.gz test.fq.gz
# 4a2e8876742302fad5bd24cba76c3cc6 C-1_R1.fastq.gz
# ea5c830f3a871b5662d63bc857ede2c2 test.fq.gz
rm test.fq.gz
Did we really change the content? Yes, because we used a different (default) compression levels to re-create the archive file.
gzip -l C-1_R1.fastq.gz
# compressed uncompressed ratio uncompressed_name
# 26042547 74702669 65.1% C-1_R1.fastq
gzip -l test.fq.gz
# compressed uncompressed ratio uncompressed_name
# 19157129 74702669 74.4% test.fq
Data Transfer
Files should be compressed (zip) or at least archived (tar) before copied or moved. It is important to verify (e.g., md5sum) data after they were moved.
Freeware
This is a personal note. We will spend a lot of money on sequencing, but not much on data analysis or software. We are used to (free) open source solutions. Remember that just because it is free does not mean that there are no restrictions. For example, most applications are free for academic use only. The code is there, but not for the taking. Freeware comes with licence agreements!
Free also does not mean free of bugs or limitations. The longer an application has been around and the larger the user community, the more people have tested it and the more likely it is that bugs have been discovered. But remember, a bug-free script still has limitations.
It is also a fact that every new approach or application released is better than any existing one, or so the authors claim. Choose your tools carefully. Always using the latest application may not be the wisest strategy.
Even a free program without a proper manual and regular maintenance is of limited use. Developers maintain tools until they are released, but often deprecate them soon after. This is a lot of work, so show your appreciation! A friendly email or tweet can go a long way. You might even consider donating to keep the tool alive.
Scripts
Have you ever wasted time trying to understand someone else's code, or struggled to find a bug because you were lost in the code? Have you ever had trouble understanding one of your older scripts? It is easy to improve your scripts. It will help you and others if you adopt a code style and use comments to explain and structure your code. Start writing code with style and be generous with comments!
Nice Code
Good developers write good code; great ones also write good comments.
Applications
It is often easier to find a solution online than to develop your own. However, there are some guidelines you should follow before frantically downloading whatever you find first. What should you consider before you click [Download]?
What to look out for?
* Copyrights / licence type * Input and output format * Understand the limitations * Check dependencies / resource requirements * Documentation * Release history
Once you have found and installed the perfect tools for your purpose. What should you do before you start processing your data?
How to get started?
* Control installation / check the logs / understand warnings * Run an example or a simplified test (e,g, dummy or toy data) * Make use of the verbose option (-v or -verbose) * Evaluate performance (e.g. speed, memory consumption) * Explore limitations
You encounter a problem with your new tool that you cannot solve. Where can you get help?
Help?
* Developers webpage * Manual / vignettes * Help option (`-help`) * ReadMe files * User community / forum * Contact developer(s)
Once you have tested the tools and successfully processed your data, you will need to give credit to the developers. What information do you need to provide to make it reproducible?
What information do I need to provide?
▻ Scripts / command lines (well commented) ▻ Version (-v, -version) ▻ Parameters / options used ▻ Reference(s)
File Exchange
There are many ways to transfer local files to or from a remote server. Choosing the right tool depends on your specific needs, preferences and environment. Command line tools such as scp or rsync are excellent for secure and automated transfers, while GUI clients such as Cyberduck or FileZilla provide a more user-friendly experience for managing files between servers and local machines.
Command-Line Tools
- scp (Secure Copy Protocol)
Transfer files securely between local and remote systems over SSH.
Usage: scp localfile.txt user@remotehost:/path/to/destination
Features: Simple syntax, secure transfer, available on most Linux-based systems.
- rsync (Remote Sync)
Synchronize files and directories between two locations over SSH.
Usage: rsync -avz localdir/ user@remotehost:/path/to/destination
Features: Incremental transfer, resume support, efficient for large transfers.
- sftp (SSH File Transfer Protocol)
Secure transfer, directory listing, and file management capabilities.
Usage: sftp user@remotehost
Features: Secure transfer, directory listing, and file management capabilities.
- wget (World Wide Web get)
Download files from the web or via FTP.
Usage: wget http://example.com/file.zip
Features: Supports HTTP, HTTPS, and FTP, can resume downloads, and download directories recursively.
- cURL (Client for URL)
Transfer data from or to a server using various protocols.
Usage: curl -O http://example.com/file.zip
Features: Supports HTTP, HTTPS, FTP, and more, highly configurable, can upload and download files.
GUI Clients
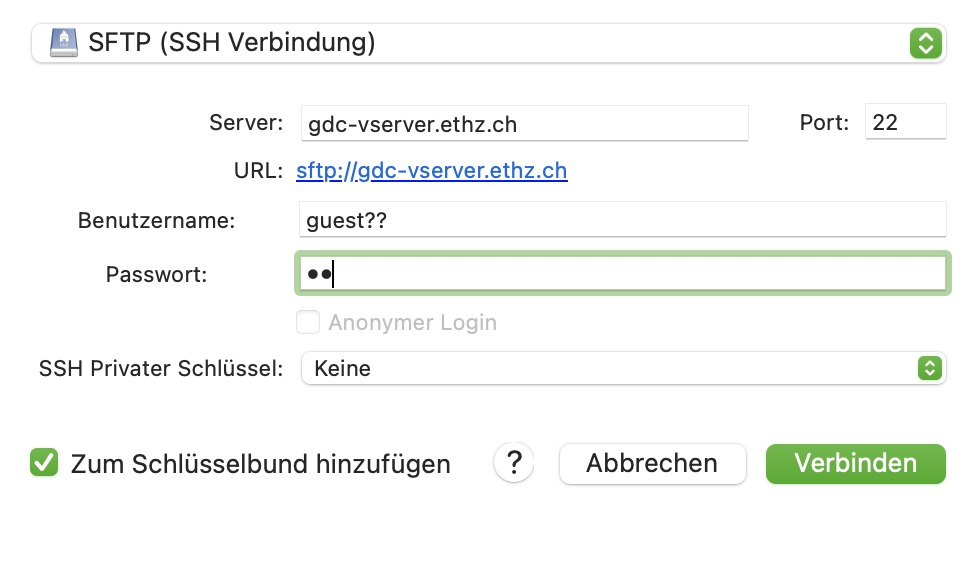
Example for Cyberduck
Settings for Cyberduck -> go to new bookmarks
Tech Terms
Source: TechTerms
Server
A server computer is a high-performance computer built to manage or store data, devices and network systems. Most servers rely on X86/X64 processors, which can run the same code as a standard desktop computer. But unlike most desktops, physical servers usually include multiple CPU sockets and error-correcting memory. The number of RAM slots on a server is generally significantly higher than on most desktop computers. The term "server" may be used for a physical machine, virtual machine (VM) or software performing server functions.
A VM is an emulated computer system. It uses physical system resources, such as the CPU, RAM, and disk storage, but is isolated from other software on the computer. It can easily be created and modified without affecting the host computer.
FTP Clients
FTP clients are used to upload, download and manage files on a (remote) server. FTP clients include, for example, Cyberduck or FileZilla.
RAID
RAID stands for “Redundant Array of Independent Disks” and is a method of storing data on multiple hard disks. When hard disks are arranged in a RAID, the computer sees them all as one big hard disk. However, they work much more efficiently than a single hard disk. The benefits of RAID come from a technique called striping, which splits up the stored data among the drives.
Shell
The shell is a purely text-based command-line interface. The user can enter commands to perform functions such as running programmes, managing directories, and displaying processes. Because the shell is only one layer above the operating system, you can perform operations that are not always possible via the graphical user interface (GUI).
VPN
VPN stands for "Virtual Private Network" and describes the possibility of establishing a protected network connection when using public networks. VPNs encrypt your internet traffic and disguise your online identity. This makes it more difficult for third parties to track your online activities and steal data. The encryption takes place in real time.
 https://3d.xkcd.com
https://3d.xkcd.com