Sequence Quality Control
Learning Objectives
◇ Understand the structure and content of FASTQ files and their significance in sequencing workflows.
◇ Use basic Bash commands to explore, inspect, and manipulate FASTQ files effectively.
◇ Comprehend the relationship between base-calling accuracy and quality scores, and what these scores indicate about sequencing data reliability.
◇ Interpret the key metrics and context-dependent visual outputs generated by FastQC to evaluate data quality.
Why Quality Control (QC) Is Essential in Sequencing Workflows
Quality control (QC) is a critical first step in any sequencing workflow. It ensures that the raw data generated by sequencing machines is reliable and free from technical artefacts that could compromise subsequent analyses. Without proper QC, common issues such as base-calling errors, adapter contamination and sequencing biases may remain unnoticed, resulting in inaccurate alignments, incorrect variant calls and, ultimately, misleading biological conclusions. By carefully assessing and filtering sequencing data at the outset, QC safeguards the integrity of the entire analysis pipeline and maximises confidence in genetic diversity results.
Base-Calling
Sequencing platforms generate raw signals as they read nucleotide sequences. Base-calling algorithms analyze these signals to determine the sequence of bases—adenine (A), thymine (T), cytosine (C), and guanine (G)—in the original DNA (or RNA) sample.
-
Illumina technology uses fluorescently labeled nucleotides and optical detection. Base calling involves interpreting the intensity and wavelength of the fluorescence signals to identify each base.
-
Pacific Biosciences (PacBio) detects fluorescently labeled nucleotides in real time during nucleotide incorporation, analyzing these signals to call bases.
-
Oxford Nanopore Technology (ONT) measures changes in ionic current as nucleotides pass through a nanopore; base calling translates these current changes into nucleotide sequences.
This translation from raw signal to nucleotide bases is inherently error-prone, and the accuracy depends heavily on the base-calling algorithm used. The confidence in each base assignment is expressed as a probability, which is encoded efficiently to reduce file size. Both the sequence and its quality probabilities are recorded together in a text-based file format called FASTQ.
FASTQ File Format
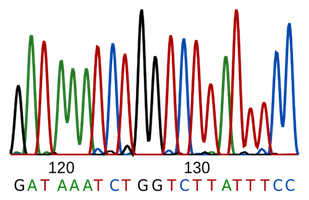
The FASTQ file format is a fixed data format that contains both the nucleotide sequence (line 2) and the corresponding quality scores (line 4).
1: @m54073_190302_105548/4325680/ccs.........................⇽ sequence header
2: TGGATCACTTGTGCAAGCATCACATCGTAGAGCTTGTCCTTGGTCTGCGA........⇽ sequence
3: +.........................................................⇽ extra line for notes
4: p~~L~jubw~n~V~2~u~[sygSN~g~K~k~~~o~u}q}|is~~~~~~~k........⇽ quality encoding
Quality Score
Quality scores based on the Phred scale are widely used to quantify the confidence in each base call made by base-calling software. These scores typically range from 0 to 40, with higher scores indicating a higher probability that the base call is correct. Mathematically, the Phred quality score Q relates logarithmically to the error probability E s follows:
where P is the probability that the base call is accurate.
Quality Encoding
To efficiently store these quality scores alongside the sequence data, they are encoded into single ASCII characters. Each quality score between 0 and 40 is converted to a corresponding ASCII symbol by adding 33 to the score’s numeric value. This encoding scheme allows compact representation of quality information in FASTQ files, with characters ranging from ! (score 0) to I (score 40).
!"#$%&'()*+,-./0123456789:;<=>?@ABCDEFGHI ➞ Encoding Character
| | | | |
34 43 53 63 73 ➞ ASCII Code
⇣ ⇣ ⇣ ⇣ ⇣
1 10 20 30 40 ➞ Q-Score (ASCII Code - 33)
⇣ ⇣ ⇣ ⇣ ⇣
21% 90% 99% 99.9% 99.99% ➞ Base call accuracy
File Size
FASTQ files can be very large - often containing millions of reads and reaching several gigabytes in size. To save storage space and improve processing efficiency, it's best to work with compressed FASTQ files (e.g., .fastq.gz). Most bioinformatics tools can read compressed files directly, so there's usually no need to decompress them.
Raw Reads Inspection
Inspecting raw sequencing reads is an essential first step in any sequencing project. Before running a full quality control (QC) report, it's important to confirm that the downloaded data is what you expect — complete, correctly paired, and uncorrupted.
A few simple command-line checks can help you:
- Confirm the file structure and completeness
- Check that forward and reverse reads are properly matched (Illumina and Aviti only)
- Identify obvious issues like truncation, missing data, or unusual headers
Catching such problems early allows you to avoid wasting time on flawed datasets, optimize your downstream analysis pipeline, and ensure your results are based on trustworthy data. This small upfront effort increases the reliability, reproducibility, and credibility of your scientific work.
A waste of time or worth your while?
It may seem like a waste of time to explore your data before you perform quality control (QC). I disagree. QC is an important step before filtering or processing data, but it has its limits. The better you understand your data from the start, the more control you have over it. For example, you have uneven sequencing depth and some samples have few reads or failed. This will help you interpret the QC report. Another example is that you notice an uneven number of reads in the R1 and R2 files. This would indicate that your download was incomplete. You discover a high level of PhiX in your subset. You are running a filter before QC. There are many good reasons to play with your data before jumping headlong into analysis.
FASTQ Case study
In this case study, we will examine a single sample that was sequenced using Illumina paired-end technology. The data consists of two compressed FASTQ files, one for each read direction.
Navigate to your working directory and verify that the expected FASTQ files are present.
## Working Directory (in case you already have it)
cd ${HOME}/GDA/qc
ls -lh C-1_R[12].fastq.gz
If the files or folder are missing, create the directory and download the sample files as follows:
# Go home
cd ${HOME}
# Create working directory
mkdir qc; cd qc
# Download sequence files
curl -O "https://www.gdc-docs.ethz.ch/GeneticDiversityAnalysis/GDA/data/C-1_R1.fastq.gz"
curl -O "https://www.gdc-docs.ethz.ch/GeneticDiversityAnalysis/GDA/data/C-1_R2.fastq.gz"
For completeness, here are the MD5 checksums of the downloaded files. You can use these to verify that the downloads are intact:
# MD5 hash for the two sequence files:
# 4a2e8876742302fad5bd24cba76c3cc6 C-1_R1.fastq.gz
# e0ce4ae266e8b2cedbc9580a3025712a C-1_R2.fastq.gz
Verification
First of all, we need to make sure that the download has worked. Using md5sum on a file you have downloaded from the internet serves several important purposes related to data integrity and security.
## Linux
md5sum C-1_R1.fastq.gz
md5sum C-1_R2.fastq.gz
## Mac
#md5 C-1_R1.fastq.gz
#md5 C-1_R2.fastq.gz
Corruption & Integrity
When downloading raw sequence read files (e.g., FASTQ files) from online databases or repositories, there is a possibility that the files could become corrupted during the transfer. To ensure data integrity, it is important to verify the downloaded files by calculating their MD5 checksum and comparing it with the MD5 hash provided by the source. This verification confirms that the files have been downloaded correctly without errors. Additionally, matching the MD5 hash guarantees that the data files are the original, unaltered versions released by the data provider, helping to prevent issues related to tampering or unintended modifications before downstream analysis.
Exploration
The following steps are flexible and not bound to a strict protocol. The key is to thoroughly examine the files or a subset of them to gain insight into their content and structure.
A good starting point is to inspect the first and last four lines of each FASTQ file. This helps verify that the sequence records appear complete and correctly formatted.
## Test Completeness
zcat C-1_R1.fastq.gz | head -n 4 # first 4 entry
zcat C-1_R1.fastq.gz | tail -n 4 # last 4 entry
zcat C-1_R2.fastq.gz | head -n 4 # first 4 entry
zcat C-1_R2.fastq.gz | tail -n 4 # last 4 entry
# Note: In case zcat does not work, try ...
# zcat < C-1_R1.fastq.gz | head -n 4
# zcat < C-1_R1.fastq.gz | tail -n 4
# or
# gzcat C-1_R2.fastq.gz | head -n 4 # first 4 entry
# gzcat C-1_R2.fastq.gz | tail -n 4 # last 4 entry
Here’s a quick reminder of what you’re looking at and what it means.
Here is a reminder of what you are looking at and what it means.
⎧the unique instrument name
| ⎧the run id
| | ⎧the flowcell id
| | | ⎧flowcell lane
| | | |
1 @M01072:41:000000000-A942B:1:1101:11853:2457 1:N:0:1
2 GTGCCAGCAGCCGCGGTAATACGTAGGTGGCAAGCGTTATCCGGATTTATTGTGC...
3 +
4 >>1>>11>11>>1EC?E?CFBFAGFC0GB/CG1EACFE/BFE///AEG1DF122A...
| | |
| | └─ C ➞ E ➞ 36 Phred Quality Score (Q) ➞ 99.975 Base call accuracy (P)
| └─ G ➞ 1 ➞ 16 Phred Quality Score (Q) ➞ 97.488 Base call accuracy (P)
└─ G ➞ > ➞ 29 Phred Quality Score (Q) ➞ 99.874 Base call accuracy (P)
Q = ascii -s ">" | awk '{print $2-33}' = 29
E = $10^{-2.9}$ = 0.001258925 ➝ 0.126%
P = $1-( 10^{-2.9})$ = 0.998741075 ➝ 99.874%
Q = ascii -s 1 | awk '{print $2-33}' = 16
E = $10^{-1.6}$ = 0.025118864 ➝ 2.511%
P = $1-( 10^{-1.6} *100)$ = 0.974881136 ➝ 97.488%
Q = ascii -s E | awk '{print $2-33}' = 36
E = $10^{-3.6}$ = 0.000251189 ➝ 0.025%
P = $100-( 10^{-3.6} *100)$ = 0.999748811 ➝ 99.975%
Results:
- The reads are 251 nucleotides in length.
- Based on the header information from the FASTQ files:
R1 head: @M01072:41:000000000-A942B:1:1101:11853:2457 1:N:0:1<br>
R2 head: @M01072:41:000000000-A942B:1:1101:11853:2457 2:N:0:1<br>
R1 tail: @M01072:41:000000000-A942B:1:2114:18740:28782 1:N:0:1<br>
R2 tail: @M01072:41:000000000-A942B:1:2114:18740:28782 2:N:0:1<br>
We can conclude the following:
- The reads are paired-end, as indicated by the matching identifiers and the 1: / 2: designation for forward and reverse reads.
- The files begin and end with matching read pairs, suggesting the data is complete and properly ordered.
- All reads originate from the same sequencing instrument, run, and flow cell, as shown by the consistent header structure.
Read Count
Next, we want to count how many sequences (reads) are in each FASTQ file. It's important to confirm that the number of forward reads (R1) matches the number of reverse reads (R2), as each pair should correspond to one DNA fragment.
In FASTA files, we count reads by counting lines that start with >. For FASTQ files, each sequence record spans four lines, with the first line (the header) starting with @. So, instead of counting >, we count lines that begin with @ — specifically, those that appear at the start of each four-line record.
To avoid decompressing the .fastq.gz files, we use zcat (or gzcat) to stream the content directly. Alternatively, zgrep can be used to search compressed files.
Count Header Lines:
zcat C-1_R1.fastq.gz | grep "@" -c
# N = 203,083
Alternative Commands
zcat < C-1_R1.fastq.gz | grep "@" -c
zgrep -c "@" C-1_R1.fastq.gz
The file C-1_R1.fastq.gz has 203,083 lines which contain a @. Does this number correspond to the number of sequence headers and can we infer the number of reads from it?
Do you see a problem with this solution?
The command counts all lines that contain @. However, not all @ lines are necessarily headers — for example, the quality line can contain @ characters.
## -----------------------------------------------------------
## Test - N(R1) == N(R2)
## We have paired-end reads (R1 and R2) and we would expect
## to have equal number or reads in both files.
## -----------------------------------------------------------
zgrep -c "@" C-1_R1.fastq.gz # N = 203,083
zgrep -c "@" C-1_R2.fastq.gz # N = 239,369
zcat C-1_R1.fastq.gz | grep "@" -c # N = 203,083
zcat C-1_R2.fastq.gz | grep "@" -c # N = 239,369
## -----------------------------------------------------------
## ASCII Table
## The ASCII character @ is also used to encode phred score 31
## and it is to be feared that we include quality encoding
## lines when counting.
## -----------------------------------------------------------
## R code - ASCII-Code Phred-Score Encoding Table
coderange <- c(33:74)
asciitable <- data.frame(coderange, coderange-33, row.names = rawToChar(as.raw(coderange), multiple = TRUE))
colnames(asciitable) <- c("ASCII","PhredScore")
asciitable
@ in the quality encoding
zcat < C-1_R1.fastq.gz | grep "?@" --color -B 3
Can you find a better (correct) solution to count the number of records in an fastq file?
## -----------------------------------------------------------
## Solution A - not really a solution :-(
## Idea: Fastq headers start with a @.
## We only count lines that start with a @.
## -----------------------------------------------------------
zgrep -c "^@" C-1_R1.fastq.gz # 145,297
zgrep -c "^@" C-1_R2.fastq.gz # 146,160
zcat C-1_R1.fastq.gz | grep "^@" -c # 145,297
zcat C-1_R2.fastq.gz | grep "^@" -c # 146,160
# ⇒ Not much better!
## -----------------------------------------------------------
## Solution B - not universal
## Idea: We extend the search query
## -----------------------------------------------------------
zcat C-1_R1.fastq.gz | head -n 1 # find unique instrument name
# @M01072:41:000000000-A942B:1:1101:11853:2457 1:N:0:1
## Solution B1
zcat C-1_R1.fastq.gz | grep "@M01072" -c # N = 144,738
zcat C-1_R2.fastq.gz | grep "@M01072" -c # N = 144,738
## Solution B2
zgrep -c "@M01072" C-1_R1.fastq.gz # N = 144,738
zgrep -c "@M01072" C-1_R2.fastq.gz # N = 144,738
# ⇒ This seems to work. Counts for both files are identical.
# ☛ This is not a universal solution.
## -----------------------------------------------------------
## Solution C
## Idea: Fastq files have 4 lines per record.
## The number of line divided by 4 = number of records
## -----------------------------------------------------------
## Solution C1
zcat C-1_R1.fastq.gz | wc -l # 578,952 / 4 = 144,738
## A few Alternatives:
# zcat < C-1_R1.fastq.gz | wc -l # for zsh
# zgrep -Ec "$" C-1_R1.fastq.gz # count lines
# gzip -cd C-1_R1.fastq.gz | wc -l # redirect gzip stdout
# ⇒ This seems to work too. Counts are identical to solution B.
# ☛ This is a universal solution but we have to some manual work.
## Solution C2
echo "$(zcat C-1_R1.fastq.gz | wc -l) / 4" | bc # N = 144,738
# echo "$(gzcat C-1_R1.fastq.gz | wc -l) / 4" | bc # zsh alternative
# bc <<< "$(wc -l < <(gzcat C-1_R1.fastq.gz)) / 4" # another one
# gzcat C-1_R1.fastq.gz | wc -l | awk '{print int($1/4)}' # one more
# ⇒ This seems to work too. Counts are identical to solution B.
# ⇒ This is a universal solution.
# ☛ It is not a fast way to count sequences.
## -----------------------------------------------------------
## Solution D
## Idea: Count lines starting (^) and ending ($) with + sign.
## -----------------------------------------------------------
zgrep -c "^+$" C-1_R1.fastq.gz # N = 144,738
# ⇒ This seems to work too and it is in agreement with B and C.
# ☛ Careful: It only works is the separator line was not altered.
We have found possible solutions to count the number of reads of a fastq file but are we sure it really works? In other words, do we really have 144'738 reads in this file?
Dummy Data
If you are not 100% sure about your command line, use a well-defined dummy dataset (e.g. extract a smaller subsample). You know what to expect, so you can test your code. You can also (double) check your results by using alternative commands/solutions. This is also a good way to broaden your command repertoire.
## Subset-Data (first 100 records only)
zcat C-1_R1.fastq.gz | head -n 400 | grep "@" -c # N = 142
zcat C-1_R1.fastq.gz | head -n 400 | grep "^@" -c # N = 100
zcat C-1_R1.fastq.gz | head -n 400 | grep "@M01072" -c # N = 100
zcat C-1_R1.fastq.gz | head -n 400 | grep -c "^+$" # N = 100
Technical Bias and Instrument ID Checking
Sequencing reads include metadata in their headers — including the instrument name, which helps trace the origin of the data. If the reads in a single FASTQ file come from multiple instruments or sequencing runs, this may indicate:
- Unintended pooling of samples
- Run-to-run variation (e.g. due to flow cell, reagents, or calibration)
- Introduction of technical bias, such as differences in error rates or quality scores
If ignored, these biases can confound downstream analysis. However, if detected early, they can be accounted for statistically (e.g. by modeling batch effects). Checking instrument IDs is a simple and effective quality control step.
Let’s inspect the FASTQ headers to check for instrument IDs.
Extract FASTQ header lines (every 4th line)
# Full header
zcat C-1_R1.fastq.gz | awk 'NR % 4 == 1' | head -n 20
# Or: strip trailing info and show only the first field of each header
zcat C-1_R1.fastq.gz | awk 'NR % 4 == 1 { print $1 }' | head -n 20
Extract only the instrument name from each header:
zcat C-1_R1.fastq.gz | awk -F":" 'NR % 4 == 1 { print $1 }' # IDs only
This works, and now we want to check if all reads came from the same instrument.
List unique instrument IDs:
zcat C-1_R1.fastq.gz | awk -F":" 'NR % 4 == 1 { print $1 }' | sort -u
This gives a list of unique IDs, but the output is limited (no counts or warnings). For a clearer summary, use the following script:
curl -O "https://www.gdc-docs.ethz.ch/GeneticDiversityAnalysis/GDA/scripts/check_ids.sh"
bash check_ids.sh <(zcat C-1_R1.fastq.gz)
If there are multiple IDs, it is important to establish whether all samples contain pooled runs or only some of them do. If only some do, we need to bear this in mind when creating downstream statistical models, including the instrument ID as a variable to test whether any observed variation can be attributed to differences between sequencing instruments.
Extract Records > Subset
A good subset is often helpful to increase processing speed when exploring your data. For example, you would like to estimate possible PhiX (an internal Illumina standard) contamination in your dataset (Mukherjee et al. 2015). Screening a subset would give you an approximate estimate, good enough to decide if a PhiX filtering step is needed.
First we would like to extract a single read.
# Get first read
zcat C-1_R1.fastq.gz | head -n 4 > First_R1.fq
# Get Last read
zcat C-1_R1.fastq.gz | tail -n 4 > Last_R1.fq
# Get a particular read
zgrep -A 3 "@M01072:41:000000000-A942B:1:1101:8091:3906" C-1_R1.fastq.gz > Exract_R1.fq
# > -A: hit and 3 lines after
# Get a random read
zgrep "@M01072" C-1_R1.fastq.gz | sort -R | head -n 1 # e.g. @M01072:41:000000000-A942B:1:1107:11792:27818 1:N:0:1
# sort: -R for random sorting
zgrep -A 3 "@M01072:41:000000000-A942B:1:1107:11792:27818 1:N:0:1" C-1_R1.fastq.gz > Random_R1.fq
# Get a random subset of reads
zgrep "@M01072" C-1_R1.fastq.gz | sort -R | head -n 10 > random10.list
zgrep -A 3 --no-group-separator -f random10.list C-1_R1.fastq.gz > Random_Subset_N10_R1.fq
However, this may not be a very fast solution. A possible faster alternative is to use seqtk to extract a random subset.
Command Not Found
The following exercises require that all necessary applications (e.g., seqtk) are installed. If you don’t have these tools locally or prefer not to install them, you can run the labs on the GDC virtual server: gdc-vserver.ethz.ch.
## Extract 10 random sequences
seqtk sample C-1_R1.fastq.gz 10 > C-1_R1_subset_10.fq
seqtk sample C-1_R2.fastq.gz 10 > C-1_R2_subset_10.fq
Compare the two subsets and pay attention to the read pairing. Can you improve the subsetting and extract the same reads from the forward (R1) and the reverse (R2) sequence file?
Can you find a way to extract a random subset of read pairs?
We use the seed option (-s[[digit]]) to keep read-pairs.
## Extract 10 random sequences with seed
seqtk sample -s123 C-1_R1.fastq.gz 10 > C-1_R1_subset_10_sync.fq
seqtk sample -s123 C-1_R2.fastq.gz 10 > C-1_R2_subset_10_sync.fq
## Compare subsets
grep "@" C-1_R1_subset_10_sync.fq | sort > R1_header.txt
grep "@" C-1_R2_subset_10_sync.fq | sort > R2_header.txt
diff R1_header.txt R2_header.txt
Find a way to extract 10 random FAST[A] sequences from C-1_R1.fastq.gz.
## -----------------------------------------------------------
## Solution A
## We only need the first two lines from a fastq record and
## we have to replace @ with >.
## -----------------------------------------------------------
## Get first two lines only (header and sequence)
zgrep -A 1 --no-group-separator -f random10.list C-1_R1.fastq.gz > Random_Subset_N10_R1.tmp
## Replace @ with >
tr "@" ">" < Random_Subset_N10_R1.tmp > Random_Subset_N10_R1.fa
## Double-check
grep ">" -c Random_Subset_N10_R1.fa
## -----------------------------------------------------------
## Solution B
## seqtk can also convert fq to fa
## -----------------------------------------------------------
seqtk seq -a C-1_R1_subset_10.fq > C-1_R1_subset_10.fa
grep ">" -c C-1_R1_subset_10.fa
Quality Control
For qualitative assessment of raw data we use FastQC. It is easy to use and produces a graphically attractive report. However, care should be taken when interpreting the results. FastQC is a contextual analysis and is designed for genomic data. When assessing quality you need to consider the type of data (e.g. RNA-Seq, Amp-Seq, RAD-Seq) and remember that a red light is not necessarily bad and a green light does not mean all is well.
## Working Directory
if [ ! -d "${HOME}/MDA/qc" ]; then
mkdir -p "${HOME}/MDA/qc"
fi
cd "${HOME}/MDA/qc"
## ---------------
## QC with FastQC
## ---------------
## Help
fastqc -help
## Version
fastqc -version
## Run FastQC on R1 and R2
fastqc -t 1 --kmers 8 C-1_R?.fastq.gz
# t number of threads
FastQC produces two output files for each sequence file. There is an interactive HTML document, and a zipped file with various outputs.
C-1_R1_fastqc.html
C-1_R1_fastqc.zip
C-1_R2_fastqc.html
C-1_R2_fastqc.zip
Use your (S)FTP client (e.g. Cyberduck) to download the HTML files and your web browser to open the report. An alternative would be to download using a terminal and the scp command. You will need to open a new (local) terminal window for this. For more information, see the "File Exchange" chapter on the Biocomputing page.
scp guest??@gdc-vserver.ethz.ch:/home/guest??/qc/C-1_R*_fastqc.html ${HOME}/qc
scp guest??@gdc-vserver.ethz.ch:/home/guest??/qc/C-1_R*_fastqc.zip ${HOME}/qc
We can also peek inside the zipped folder and open the text-based reports.
## Explore the zipped Results
less C-1_R1_fastqc.zip
unzip -p C-1_R1_fastqc.zip C-1_R1_fastqc/summary.txt | less
FastQC has a range of interesting options. On the one hand, you can load zipped Fastq files, but other data formats can also be used (e.g. BAM, fast5). You can also search for costum adapters or target sequences (e.g. contaminants).
QC Facts & Considerations
There are a few things you might consider bevore running a quality control.
➼ Subset - Does it make sense to run QC on the entire dataset? Would a subset also work?
➼ Data Structure - QC is based on data structure analysis. You have to know the structure of your data.
➼ Adaptors - Make sure you are looking for the right adaptor sequences.
➼ Quality Drop - The quality decreases with read length.
➼ R1 vs. R2 - Average quality is higher for the first (forward) read (R1) compare to the second (reverse) read (R2).
RTFM (Read The Fine Manual)
Like all applications, FastQC has limitations and settings that the user needs to be aware of. For this reason I think it is better to know a few applications well than many superficially. Reading the manual is a good starting point to understand an application better. I agree with you. Some manuals (but not all!) are terrible to read. Sentences like "use the settings button to change the settings" are not very helpful, but unfortunately common. But at least give each manual a chance, because the author(s) have spent some time on it. The FastQC manual (link below) is an example of a helpful manual. You will learn that some modules (e.g. Duplicate Sequences) use "shortcuts" to increase the speed of analysis. It is important that you know and understand these limitations.
FastQC with R
Windows and FastQC in R
If you use a Linux emulator FastQC for R does not work. Rqc would be a possible alternative (see below). If you have Ubuntu installed you have to specify the path to the installation.
## Install / Load fastqcr
#install.packages("fastqcr")
library("fastqcr")
## FastQC
fastqc(fq.dir = "${HOME}/qc/", qc.dir = "${HOME}/qc/"))
## FastQC for Windows with Ubuntu
# fastqcr::fastqc("seq.fq.gz", fastqc.path = "wsl.exe /path_to_wsl_fastqc_install/fastqc")
## QC-Plots
qc.C01.R1 <- qc_read(file = "~/qc/C-1_R1_fastqc.zip", modules = "all", verbose = TRUE)
qc_plot(qc.C01.R1, "Per sequence GC content")
qc_plot(qc.C01.R1, "Per base sequence quality")
qc_plot(qc.C01.R1, "Per sequence quality scores")
qc_plot(qc.C01.R1, "Per base sequence content")
qc_plot(qc.C01.R1, "Sequence duplication levels")
## Aggregate FastQC Reports for Multiple Samples
qc <- qc_aggregate(qc.dir = "~/qc/")
## Summary Report
library(dplyr)
qc %>%
select(sample, module, status) %>%
filter(status %in% c("WARN", "FAIL")) %>%
arrange(sample)
## Generate Multi QC Report
qc_report(qc.path = "~/qc/", result.file = "/Users/jwalser/qc",
experiment = "GDA - Test")
More R Packages for QC
Rqc is an optimized tool designed for quality control and assessment of high-throughput sequencing data. It performs parallel processing of entire files and produces a report which contains a set of high-resolution graphics that can be used for quality assessment.
library(Rqc)
# Depends on: BiocParallel, ShortRead, ggplot2
library(ggplot2)
library(BiocParallel)
library(ShortRead)
Rqc::rqc(path = "/path/to/workfolder", pattern = ".fastq.gz")
# Basic Usage:
# rqc(path = folder, pattern = ".fastq.gz")
# folder: folder with squences
# pattern: regular expression that identifies all files of interest
QC Summary
With MultiQC it is possible to combine individual FastQC reports into one interactive report in HTML format.
QC Screening
FastQ Screen allows you to compare fastq sequences to a number of sequence databases to better understand library composition.
Additional information
Literature
- Mukherjee et al. (2015) Large-scale contamination of microbial isolate genomes by Illumina PhiX control
- Understanding Illumina Quality Scores