Beta Diversity Help¶
A quick summary to better understand beta diveristy. To make this work, you need to download and import the dummy dataset described in chapter ”Phyloseq Help”.
Compositional Beat-Diversity¶
Help with Distance¶
distanceMethodList betadiver(help = TRUE) help(designdist) # designdist {vegan}
We use different OTU-abundance-distribution scenarios to better understand Beta-diversity indices.
### Example [A]: Frequent OTU overlap - rare ones are unique ## Explore data t(otu_table(d.B)) ## Wide-to-long format - ideal for ggplots! x <- data.frame(otu_table(d.B)) x$OTU <- row.names(x) x.new <- gather(x, Sample, Counts, SB1, SB2) ## Barplot # Sample order - oneway or another # position <- gtools::mixedsort(x$OTU) position <- c("OTU1","OTU2","OTU3","OTU4","OTU5","OTU6","OTU7","OTU8","OTU9","OTU10","OTU11","OTU12","OTU13","OTU14","OTU15") p <- ggplot(x.new, aes(x = factor(OTU, levels = position ), y = Counts, fill = factor(Sample))) + geom_bar(stat = "identity", position = "dodge") + xlab("OTU") + ylab("Counts") p
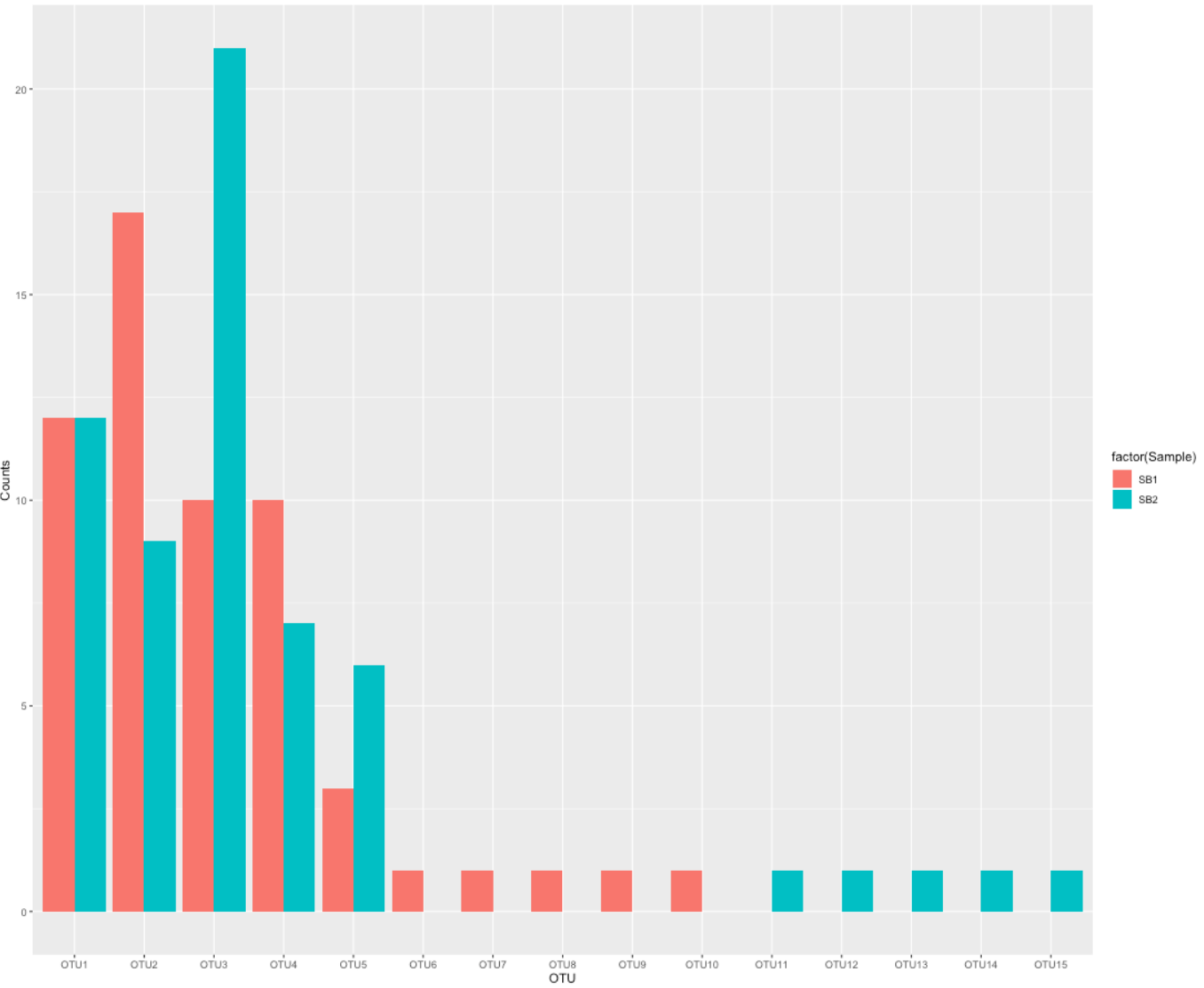
We create a few more example cases and record the distances in a data frame:
## Create empty data frame df <- setNames(data.frame(matrix(ncol = 4, nrow = 5)), c("Bray-Curtis (b)", "Jaccard (b)","Bray-Curtis (q)", "Jaccard (q)")) row.names(df) <- c("Frequent-Overlap", "Frequent-Overlap(equal)", "Rare-Overlap", "Identical", "Different") ## Binary dist.bc.b <- phyloseq::distance(d.B, method = "(A+B-2*J)/(A+B)") ## "minimum" Bray-Curtis dist.jc.b <- phyloseq::distance(d.B, method = "cc") ## Jaccard index of dissimilarity dist.jc.b <- phyloseq::distance(d.B, method = "jaccard", binary = TRUE) ## vegdist jaccard ## Quantitative dist.bc.q <- phyloseq::distance(d.B, method = "bray") ## Bray-Curtis dist.jc.q <- phyloseq::distance(d.B, method = "jaccard") ## Jaccard index of dissimilarity df[1,1] <- dist.bc.b df[1,2] <- dist.jc.b df[1,3] <- dist.bc.q df[1,4] <- dist.jc.q ### Example [B]: Frequent Overlap ## Change counts by hand otu_table(d.B)[,1] <- c(20,10,10,5,5,1,1,1,1,1,0,0,0,0,0) otu_table(d.B)[,2] <- c(20,10,10,5,5,0,0,0,0,0,1,1,1,1,1) t(otu_table(d.B)) ## Binary dist.bc.b <- phyloseq::distance(d.B, method = "(A+B-2*J)/(A+B)") ## "minimum" Bray-Curtis dist.jc.b <- phyloseq::distance(d.B, method = "cc") ## Jaccard index of dissimilarity ## Quantitative dist.bc.q <- phyloseq::distance(d.B, method = "bray") ## Bray-Curtis dist.jc.q <- phyloseq::distance(d.B, method = "jaccard") ## Jaccard index of dissimilarity df[2,1] <- dist.bc.b df[2,2] <- dist.jc.b df[2,3] <- dist.bc.q df[2,4] <- dist.jc.q ### Example [C]: Rare Overlap ## Change counts by hand otu_table(d.B)[,1] <- c(20,10,10,5,5,1,1,1,1,1,0,0,0,0,0) otu_table(d.B)[,2] <- c(0,0,0,0,0,1,1,1,1,1,5,5,10,10,20) t(otu_table(d.B)) ## Binary dist.bc.b <- phyloseq::distance(d.B, method = "(A+B-2*J)/(A+B)") ## "minimum" Bray-Curtis dist.jc.b <- phyloseq::distance(d.B, method = "cc") ## Jaccard index of dissimilarity ## Quantitative dist.bc.q <- phyloseq::distance(d.B, method = "bray") ## Bray-Curtis dist.jc.q <- phyloseq::distance(d.B, method = "jaccard") ## Jaccard index of dissimilarity df[3,1] <- dist.bc.b df[3,2] <- dist.jc.b df[3,3] <- dist.bc.q df[3,4] <- dist.jc.q ### Example [D]: Both identical ## Change counts by hand otu_table(d.B)[,1] <- c(20,10,10,5,5,1,1,1,1,1,0,0,0,0,0) otu_table(d.B)[,2] <- c(20,10,10,5,5,1,1,1,1,1,0,0,0,0,0) t(otu_table(d.B)) ## Binary dist.bc.b <- phyloseq::distance(d.B, method = "(A+B-2*J)/(A+B)") ## "minimum" Bray-Curtis dist.jc.b <- phyloseq::distance(d.B, method = "cc") ## Jaccard index of dissimilarity ## Quantitative dist.bc.q <- phyloseq::distance(d.B, method = "bray") ## Bray-Curtis dist.jc.q <- phyloseq::distance(d.B, method = "jaccard") ## Jaccard index of dissimilarity df[4,1] <- dist.bc.b df[4,2] <- dist.jc.b df[4,3] <- dist.bc.q df[4,4] <- dist.jc.q ### Example [E]: Nothing in common ## Change counts by hand otu_table(d.B)[,1] <- c(20,10,10,5,5,1,1,1,0,0,0,0,0,0,0) otu_table(d.B)[,2] <- c(0,0,0,0,0,0,0,0,1,1,5,5,10,10,20) t(otu_table(d.B)) ## Binary dist.bc.b <- phyloseq::distance(d.B, method = "(A+B-2*J)/(A+B)") ## "minimum" Bray-Curtis dist.jc.b <- phyloseq::distance(d.B, method = "cc") ## Jaccard index of dissimilarity ## Quantitative dist.bc.q <- phyloseq::distance(d.B, method = "bray") ## Bray-Curtis dist.jc.q <- phyloseq::distance(d.B, method = "jaccard") ## Jaccard index of dissimilarity df[5,1] <- dist.bc.b df[5,2] <- dist.jc.b df[5,3] <- dist.bc.q df[5,4] <- dist.jc.q round(df, 3)
| Bray-Curtis(b) | Jaccard(b) | Bray-Curtis(q) | Jaccard(q) | |
|---|---|---|---|---|
| Frequent-Overlap | 0.5 | 0.667 | 0.299 | 0.461 |
| Frequent-Overlap(equal) | 0.5 | 0.667 | 0.091 | 0.167 |
| Rare-Overlap | 0.5 | 0.667 | 0.909 | 0.952 |
| Identical | 0.0 | 0.000 | 0.000 | 0.000 |
| Different | 1.0 | 1.000 | 1.000 | 1.000 |
Phylogenetic Beta Diversity¶
Note
Careful with data manipulations. It is possible that even simple manipulations change the structure of the object or parts of it. For example, subsetting the imported data resulted in a change of the phylognetic tree information of the phyloseq object.
phy_tree(dB) # tree is rooted phy_tree(d.B) # tree is unrooted distance(dB, method = "unifrac") # rooted tree distance(d.B, method = "unifrac") # unrooted tree
To avoid that a random root is selected we have to re-root the tree by hand.
## Re-root the tree old.tree <- phy_tree(d.B) new.tree <- ape::root(old.tree, outgroup = "OTU13", resolve.root = TRUE) phy_tree(d.B) <- new.tree phy_tree(d.B)
To better understand beta diversity based on phylogenetic information, we create different dummy datasets and compare distances calculated with Unifrac and Weighted Unifrac.
# (A) All Equal otu_table(d.B)[,1] <- c(1,1,1,1,1,1,1,1,1,1,1,1,1,1,1) otu_table(d.B)[,2] <- c(1,1,1,1,1,1,1,1,1,1,1,1,1,1,1) distance(d.B, method = "unifrac") ## Unifrac distance(d.B, method = "wunifrac") ## Weighted Unifrac plot_tree(d.B, label.tips = "taxa_names", color = "SampleID", size = "Abundance") # (B) All Different otu_table(d.B)[,1] <- c(1,1,1,1,1,0,0,0,0,0,0,0,0,0,1) otu_table(d.B)[,2] <- c(0,0,0,0,0,1,1,1,1,1,1,1,1,1,0) distance(d.B, method = "unifrac") ## Unifrac distance(d.B, method = "wunifrac") ## Weighted Unifrac plot_tree(d.B, label.tips = "taxa_names", color = "SampleID", size = "Abundance") # (C) Mixed taxa with difference abundance otu_table(d.B)[,1] <- c(10,1,10,1,10,1,10,1,10,1,10,1,10,1,1) otu_table(d.B)[,2] <- c(1,10,1,10,1,10,1,10,1,10,1,10,1,10,10) distance(d.B, method = "unifrac") ## Unifrac distance(d.B, method = "wunifrac") ## Weighted Unifrac plot_tree(d.B, label.tips = "taxa_names", color = "SampleID", size = "Abundance") # (D) Mixed taxa at low abundance otu_table(d.B)[,1] <- c(1,0,1,0,1,0,1,0,1,0,1,1,1,1,1) otu_table(d.B)[,2] <- c(0,1,0,1,0,1,0,1,0,1,0,1,1,1,1) distance(d.B, method = "unifrac") ## Unifrac distance(d.B, method = "wunifrac") ## Weighted Unifrac plot_tree(d.B, label.tips = "taxa_names", color = "SampleID", size = "Abundance") # (E) Mixed taxa at higher abundance otu_table(d.B)[,1] <- c(10,0,10,0,10,0,10,0,10,0,10,10,10,10,10) otu_table(d.B)[,2] <- c(0,10,0,10,0,10,0,10,0,10,0,10,10,10,10) distance(d.B, method = "unifrac") ## Unifrac distance(d.B, method = "wunifrac") ## Weighted Unifrac plot_tree(d.B, label.tips = "taxa_names", color = "SampleID") # (F) A few distinct taxa at higher abundance otu_table(d.B)[,1] <- c(1,1,1,1,1,1,1,1,1,1,1,10,10,10,1) otu_table(d.B)[,2] <- c(1,1,1,1,1,1,1,1,1,1,1,0,0,0,1) distance(d.B, method = "unifrac") ## Unifrac distance(d.B, method = "wunifrac") ## Weighted Unifrac plot_tree(d.B, label.tips = "taxa_names", color = "SampleID", size = "Abundance") # (G) A few distinct taxa at lower abundance otu_table(d.B)[,1] <- c(100,1,1,1,1,1,1,1,1,1,1,10,10,10,1) otu_table(d.B)[,2] <- c(100,1,1,1,1,1,1,1,1,1,1,0,0,0,1) distance(d.B, method = "unifrac") ## Unifrac distance(d.B, method = "wunifrac") ## Weighted Unifrac plot_tree(d.B, label.tips = "taxa_names", color = "SampleID", size = "Abundance")