Biocomputing on a HPC cluster
In contrast to a server (e.g. our GDC severs) a high performance computing (HPC) cluster (e.g. Euler or Leonhard) contains many thousands of inter-connected compute nodes. You can thus use much more resources, but you need to consider some aspects that we will discuss here. For this tutorial we will virtually work on Euler and there is no access needed. Every ETH student can of course login and try some of the commands. Students from unizh have their own cluster, which might be different. However, the points discussed here might be also relevant.
Finde more information to Euler and Leonhard on the official Euler wiki.
More specific information is provided in the GDC Euler manual.
Basic terms¶
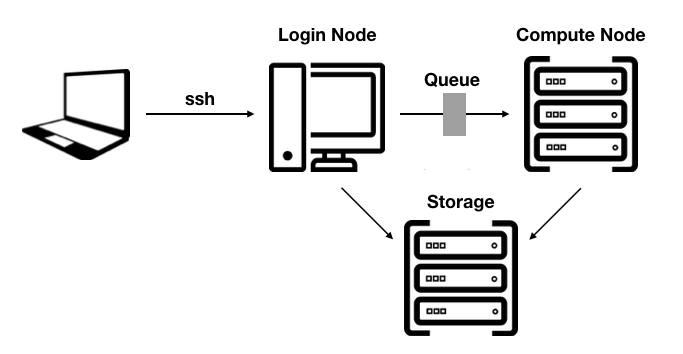
- HPC cluster: relatively tightly coupled collection of compute nodes. Access to the cluster is provided through a login node. A resource manager and scheduler provide the logic to schedule jobs efficiently on the cluster.
-
Login node: Serve as an access point for users wishing to run jobs on the HPC cluster. Do not run demanding jobs on the login nodes.
-
Compute node: Currently most compute nodes have two sockets, each with a single CPU, volatile working memory (RAM), a hard drive, typically small and only used to store temporary files, and a network card.
-
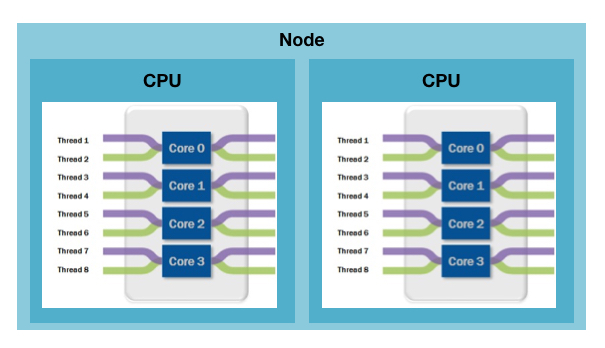
CPU: Central Processing Unit, the chip that performs the actual computation in a compute node. A modern CPU is composed of numerous cores, typically 8 or 10. It has also several cache levels that help in data reuse.
-
Core: part of a modern CPU. A core is capable of running processes and has its own processing logic and floating point unit. Each core has its own level 1 and level 2 cache for data and instructions. Cores share last level cache.
-
Threads: a process can perform multiple computations, i.e. program flows, concurrently. In scientific applications threads typically process their own subset of data or a subset of loop iterations
Quote
What is the difference between a core and a thread? Think of the core as a person’s mouth and the threads as the hands. The mouth does all of the eating, while the hands just help organise the ‘workload’. The thread helps deliver the workload to the CPU more efficiently. More threads translates into a better-organized work queue, hence more efficiency in processing the information (https://www.techsiting.com/cores-vs-threads/).
- Memory: Each processor needs memory associated with it to provide a place for the processor to do its work. Some applications (e.g. genome assemblies) needs a lot of memory. On Euler/Leonhard memory is limited as it's compared to CPUs expensive and thus you need to optimise your jobs.
The batch system, called LSF (Load Sharing Facility), manages and schedules your job(s). In order to use the cluster, you need to define the resources (CPUs, memory) and the run-time in advance.If you are requesting not sufficant ressouces the jobs gets killed by LSF. If you are requesting too many resources, CPUs idle and memory gets wasted. Thus, requested resources (CPU, memory or run-time) of your jobs needs to be optimised.
What do I need to know to run an optimized job?
How long the job will run ?
How much memory the job will use ?
How many CPU-cores you need ?
Important
The better your jobs are optimized, the more you can run at the same time and the faster you can finish the analysis.
Tipp
As a general rule, you should only request > 4 CPUs per job if you know that the program scales well. Many bioinformatic tools do not scale anymore at some point.
Tipp
Avoid to script long pipelines which use many different tools with different memory or CPU requirements. Often one cannot optimise entire pipelines as for example a lot of memory is only used in a certain stage. Please use job chaining or run the different tools independently, where you can optimise the resources much easier. It is also less error prone.
Challenges¶
(1) You going to use an unknown tool. How do you know how much memory, CPUs and time do you need?
Suggestion
You need to run some tests (e.g. a part of your jobs or reduced dataset) and monitore the jobs constantly in order to find the optimal settings for your jobs. The better the jobs are optimised the more jobs you can run in parallel. If the test jobs get killed by the system increase the resources eventually. Do not request by default > 30 Gb as it is a lot and limited on Euler.
(2) Your tool seems to run only on 1 CPU (R scripts or I found no evidence for using multiple CPUs in the manual). Does it run faster if I request 4 CPUs?
Suggestion
If you can thread a tool or not depends on its architecture. Thus you cannot speed up a tool by just requesting more CPUs.
(3) Your tool seems to use 4 Gb memory. Does it runs faster if I request 8 Gb?
Suggestion
Normally an increase of memory does not speed up your job except the tool have such function implemented.
(4) You like to run fastqc on multiple CPUs and you know from the manual that it is possible. Does the tool uses by default multiple CPUs?
Suggestion
Normally not make sure that you are define the number of CPUs using e.g. the -t option for fastqc.
(5) You job has been killed because of exceeding memory limits. Does it make sense to just request more CPUs (e.g. increase the total of requested memory)?
If memory is limiting you need to increase the total amount of memory or use the same amount but request less CPUs. The ratio memory to CPUs is decisive.
Suggestion
Cluster related commands¶
Overview of the submitted jobs
bjobs bjobs <job-ID>
CPU and memory usage of running jobs
bbjobs bbjobs <job-ID>
Summary of all submitted jobs
busers
Kill specific job
bkill <job-ID>
Kill all running jobs
bkill 0
Kill job 15-23 of job array job-ID
bkill <job-ID>[15-23]
File system¶
On a HPC cluster you have often several file systems. On Euler/Leonhard there is your personal Euler home directory (/cluster/home/<USER>), where you can keep scripts or own installed tools. Your personal scratch folder (/cluster/scratch/<USER>) is your working directory. Data will be automatically deleted after two weeks. Thus important files have to be copied to "work" or "project" for save-keeping. Always make sure that there is enough disk space available on your target volume.
Challenges¶
(1) You received 200 G bp gzipped fastq files. Should I unzip the files?
Suggestion
Never extract fastq files, remember they will get roughly 4 times larger (~800 Gb). Most of the tools can handle the compressed files.
(2) You will produce 100,000 of small files from simulations. Can I output the data directly to "work" or "project"?
Suggestion
Use the scratch disks for this task and keep only the final data. If you like to keep all the files compress the entire folder and copy only the tar file to "work,"projects" or any other mounted volumes.
Software pool¶
As so many software tools are installed on Euler they can interfere with each other, e.g. they need different compilers or run-time libraries. Therefore, software tools have to be "loaded" individually before they can be used. This will make sure that the correct libraries and paths will be available.
Here you find a list of all installed tools installed in the GDC stack.
Challenge¶
(1) Which modules do I need to load to use bwa version 0.7.17 ?
Suggestion
module load gcc/4.8.2 gdc bwa/0.7.17
(2) Which modules do I need to load to use gatk version 4.1.5 ?
Suggestion
This version is not installed in the GDC software stack. Use another version.
Job submission¶
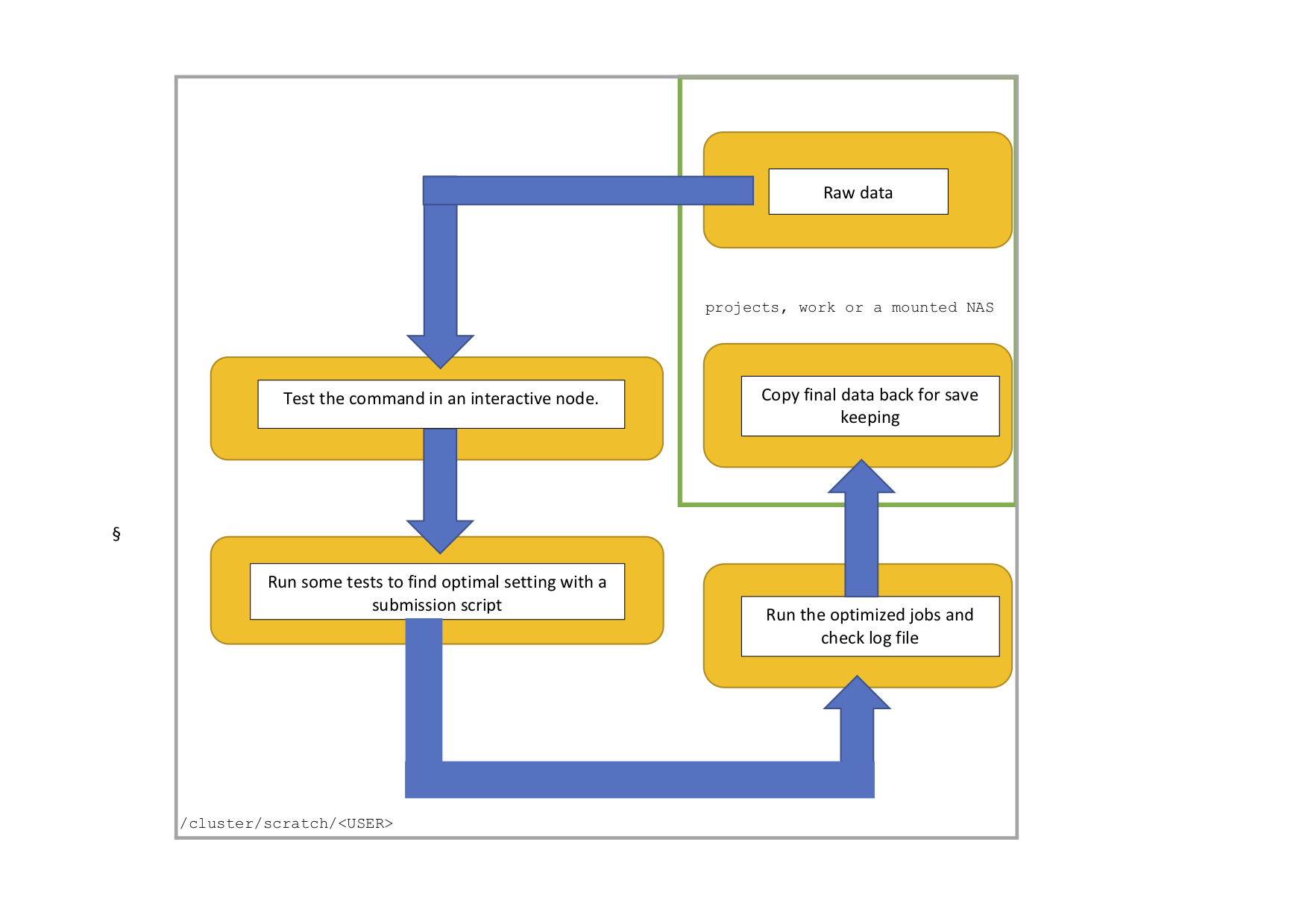
This is a typical workflow on Euler. Your working directory is the scratch, the raw data is stored on projects, work or other mounted NAS. In an interactive node you can then test the command. In the next step you run some test runs using a submission script to find the optimal resources (CPU, memory and time). Information about running jobs can be obtained through the monitoring tools, as explained below. Afterwards you can run all jobs, check the log file and transfer only the output that you like to keep to your project, work or mounted NAS space.
Interactive node¶
If you like to work on a cluster like a normal server and get the outputs directly in the terminal e.g. grep some patterns or you like to test a command, use an interactive node.
bsub -W 4:00 -Is bash
Single job¶
For reproducible research it is important to use scripts rather than just commands. We now going to do a joint SNP calling using FreeBayes.
#!/bin/bash #BSUB -J "freebayes" #Name of the job #BSUB -R "rusage[mem=10000]" #Requesting 10 Gb memory per core, in total 20 G #BSUB -n 2 #Requesting 2 core #BSUB -W 24:00 #Requesting 4 hours running time #Load the needed tools module load gcc/4.8.2 gdc freebayes/1.0.2 #The freebayes command, for a list of bam files freebayes -L bam.list -v out.vcf –f ref.fas
The script (name: submit-freebayes-single_job.lsf) that you would prepare in an editor includs some explanations. The actual FreeBayes command is on the last line. This is the command you would run on the command-line of a regular server.
To run this submission script, you would type:
bsub < submit-freebayes-single_job.lsf.sh
Challenge¶
1 CPU, 8 Gb and 10 hours seems to be sufficient for the FreeBayes based on some test runs. You have also realised that the bam list was wrong, the correct name is bam_v2.list. Updated the submission script.
Suggestion
#!/bin/bash #BSUB -J "freebayes" #Name of the job #BSUB -R "rusage[mem=8000]" #Requesting 8 Gb memory per core #BSUB -n 1 #Requesting 1 core #BSUB -W 12:00 #Requesting 12 hours running time #Load the needed tools module load gcc/4.8.2 gdc freebayes/1.0.2 #The freebayes command, for a list of bam files freebayes -L bam_v2.list -v out.vcf –f ref.fa
Job array¶
Many computing tasks can be split up into smaller tasks that can run in parallel (e.g BLAST, read mapping, SNP-calling). A cluster is extremely well suited for such jobs and the bsub command has an option specifically for this.
Let's do the mapping of 20 samples using BWA.The sample names are provided in a list and we iterate (similar to a loop in the SNP challenge) over the samples using a job array.
#!/bin/bash #BSUB -J "bwa[1-20]%10" #Array with 20 Jobs, always 10 running in parallel #BSUB -R "rusage[mem=4000]" #Requesting 4 Gb per sub job, in total 8 Gb #BSUB -n 2 #Requesting 2 core per sub job #BSUB -W 4:00 #4 hours run-time per sub job #Load modules module load gcc/4.8.2 gdc bwa/0.7.12 ##provide path in=samples out=mapping ##generate output folder if not present if [ ! -e ${out} ] ; then mkdir ${out} ; fi #Internal job index variable (length 20) see [1-20] IDX=$LSB_JOBINDEX #Extract the sample names from a list (sample.list) based on the number of lines and save it in the variable sample sample=$(sed -n ${IDX}p <sample.list) #Run BWA bwa mem Ref.fasta ${in}/${sample}_R1.fq.gz ${in}/${sample}_R2.fq.gz -t 2 > ${out}/${sample}.sam
To run this submission script, you would type:
bsub < submit-bwa-array_job.lsf.sh
Challenge¶
In total 12 Gb memory seems to be sufficant for each job. You have another 300 samples and like to run 48 CPUs in total. Each job you like to run on 4 CPUs. The fastq files are located in the folder raw and you like to output the sam files to a folder called mapping2. Update the submission script.
Suggestion
#!/bin/bash #BSUB -J "bwa[1-300]%12" #Array with 300 Jobs, always 12 running in parallel #BSUB -R "rusage[mem=3000]" #Requesting 3 Gb per sub job, in total 12 Gb #BSUB -n 4 #Requesting 4 core per sub job #BSUB -W 4:00 #4 hours run-time per sub job #Load modules module load gcc/4.8.2 gdc bwa/0.7.12 #Internal job index variable (length 300) IDX=$LSB_JOBINDEX ##provide path in=raw out=mapping2 ##generate output folder if not present if [ ! -e ${out} ] ; then mkdir ${out} ; fi #Internal job index variable (length 300) IDX=$LSB_JOBINDEX #Extract the sample names from a list (sample.list) sample=$(sed -n ${IDX}p <sample.list) #Run BWA bwa mem Ref.fasta ${in}/${sample}_R1.fq.gz ${in}/${sample}_R2.fq.gz -t 4 > ${out}/${sample}.sam
R scripts¶
Running Rstudio on HPC clusters can be sometimes a bit annoying, especially if you like to visualise your data. If you have entire workflows you can submit Rscripts via bsub, save the data as Rdata and do e.g. the plotting or the filtering locally on your computer. Here an example of an R script that you use for just read a huge data file and save it as a Rdata object.
Let's copy the lines below in a file and call it Reformat.R.
#!/usr/bin/env Rscript ## Use this argument to provide file names in the command below, order must be consistent args <- commandArgs(trailingOnly=TRUE) library(tidyverse) ## Read table samples <- read_csv(args[1], header = FALSE) ## Save it as a RData file name<- paste(args[2], "RData") save(samples, name)
As we normally don't want to load user specific data we use --vanilla option.
bsub -n 1 -W 4:00 "Rscript --vanilla Reformat.R Fst_chromosomes1.txt Fst_chromosomes1_reduced"
This command will then output the file Fst_chromosomes1_reduced.RData
Challenge¶
You have realised that there is a header in the table and that the file name is Pi_chromosomes1_reduced. Update the script accordingly.
Suggestion
#!/usr/bin/env Rscript ## Use this argument to provide file names in the command below, order must be consistent args <- commandArgs(trailingOnly=TRUE) library(tidyverse) ## Read table samples <- read_csv(args[1], header = TRUE) ## Save it as a RData file name<- paste(args[2], "RData") save(samples, name)
bsub -n 1 -W 1:00 "Rscript --vanilla Reformat.R Pi_chromosomes1.txt Pi_chromosomes1_reduced"
Optimization¶
Before you submit a batch of jobs you have to optimise the resource request. This might be difficult but there are tools like bbjobs and the lsf output file might help you.
Quote
bbjobs is your best friend (Nik Zemp).
We would like to do a SNP calling and have split the reference genome in 120 chunks. Now we need to find out how many CPU and how much memory is needed for each of the 120 jobs.
(1) Run a test with 5 representative chunks (do not just take the first 5) with 4 CPUs and 4X20 Gb RAM and 24 hours run time.
#!/bin/bash #BSUB -J "freebayes[1, 10, 15, 20, 50]" #Running job 1,10,15,20,50 #BSUB -R "rusage[mem=20000]" #Requesting 4 X 20 Gb per job #BSUB -n 4 #Requesting 4 core per job array sub-job #BSUB -W 24:00 #24 hours run-time per job
(2) After 30 min the CPU as well as the memory usage is pretty low using bbjobs.
CPU utilization: 24% Resident memory utilization: 20%
(3) Let's restart the 5 representative chunks (do not just take the first 5) with 2 CPUs and 2 X 20 Gb memory and 24 hours run time.
#!/bin/bash #BSUB -J "freebayes[1, 10, 15, 20, 50]“ #Running job 1,10,15,20,50 #BSUB -R "rusage[mem=20000]" #Requesting 2 X 20 Gb per job, 40 Gb in total #BSUB -n 2 #Requesting 2 core per job array sub-job #BSUB -W 24:00 #24 hours run-time per job
(4) After your runs are finished please also have a look at the lsf.o-file. In particular "Max Memory" and "Run time" are important:
CPU time : 3025 sec.
Max Memory : 30000 MB
Average Memory : 25000 MB
Total Requested Memory : 40000 MB
Delta Memory : 10000 MB
Max Swap : -
Max Processes : 4
Max Threads : 9
Run time : 2995 sec.
Turnaround time : 20998 sec.
In this example the run time is 2995 sec. The memory usage is 75% (Max. Memory/Total Requested Memory). Of the requested 2 CPUs only about 1 (Run time/CPU time) were used. Ideally, CPU time ~ number of CPUs times run time.
(5) Let’s have a look at the other runs. 4 out of the 5 runs had similar usage. One of the jobs was killed (20%) because there was not enough memory available. There will often be some jobs that get killed when using tools with rather large variance in resource usage. But the proportion is small so we rerun the failed jobs later.
Challenge¶
(6) What would you now request for all 120 jobs?
Suggestion
1 CPU and 1x30 Gb RAM for 4 hours
#!/bin/bash #BSUB -J "freebayes[1-120]%20" #Running job 1-120, 20 in parallel #BSUB -R "rusage[mem=30000]" #Requesting 1 X 30 Gb per job #BSUB -n 1 #Requesting 1 core per job array sub-job #BSUB -W 4:00 #4 hours run-time per job
(7) After all jobs are finished you can check which jobs have failed and rerun them again with 1 X 60 Gb memory.
#!/bin/bash #BSUB -J "freebayes[1, 10, 17-20, 51]" #Rerun the failed jobs based on the log file #BSUB -R "rusage[mem=60000]" #Requesting 1 X 60 Gb per job #BSUB -n 1 #Requesting 1 core per job array sub-job #BSUB -W 4:00 #4hours run-time per job