Next Generation Sequencing - Introduction¶

Lecture Notes¶
Rapid advancements in modern DNA sequencing technologies have (and still does) revolutionize biological and medical science.
"The major factors, such as increasing applications of NGS, speed, cost, and accuracy, efficient replacement for traditional technologies, and drug discovery applications demanding NGS technology are expected to drive the growth of the overall market." (ResearchAndMarkets.com)
The ease to produce data has shifted the principal research focus from loci to genomes, increased sample numbers, requires data management strategies, and changed data analysis. As a result, nucleotide sequence archives (ENA, NCBI) are growing daily.
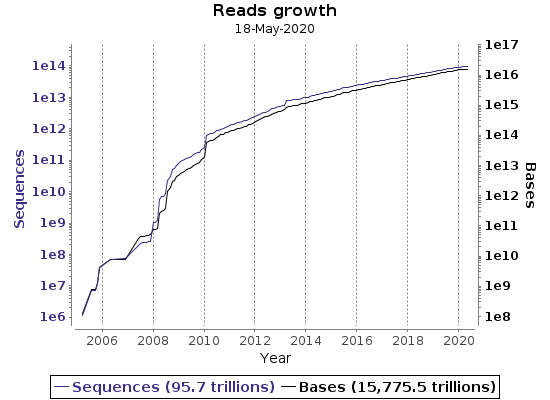
ENA regularly collectis several statistics regarding data growth.
Unfortunately, the high-throughput-sequencing-hype has also removed the research question and researchers have started to sequences everything just because we can. It is paramount to understand the pros and cons of each technology to optimise the sequencing design according to the question(s) and resources at hand.
Sequencing Technology Explained¶
The suggested movies below will help you better understand the different sequencing technologies. The selection is by no means comprehensive, but it will cover the basics.
- Sanger Sequencing
- Sanger Sequencing by Thermo Fisher Scientific
- Roche 454 - Music only
- 454 Sequencing - Explained
- Ion Torrent
- Illumina Sequencing Technology
- MGI
- PacBio SMRT Sequencing
- ONT Nanopore Sequencing
- BioNano Genomics Irys Technology
Comparison¶
There are many detailed reviews available that compare the different NGS technologies. The problem, they are outmoded the moment they are published. Sequence technology is moving fast and it is difficult to keep up with the latest development. Following a more general and personal comparison as an approximate guide to NGS.
| Platfom | Technology | Data | Template | DNA | Lib-Prep | Indexing | Length | Error |
|---|---|---|---|---|---|---|---|---|
| Illumina | established | lots | low-high | all | easy | easy | short | moderate |
| PacBio | established | less | high | high-mol | difficult | difficult | long | high-low |
| ONT | experimental | less | higher | high-mol | easy | difficult | long | high |
The table does not include all technonogies but only the ones the GDC most often handels. The table also ignores solutions for specific applications. We at the GDC believe that the question and all the resources available (not only money) should determin the sequencing technology. There is no point of aiming for long reads if you have not enough and/or highly fragmented starting DNA.
NGS Sequence File Format (Raw Data)¶
Illumina sequence reads are usually provided as de-multiplexed fastq (i.e. fastq.gz) files. PacBio sequences reads are either provided as fastq, circular consensus sequences (ccs) fasta or BAM files. CCS files can be considered error-corrected sequence reads. Shorter sequence fragments are read multiple time (circled) during a PacBio run and with each pass the accuracy increases. The BAM file can be converted to fastq format using the PacBio BAM recipes. Oxford Nanopore (ONT) raw data are provided as fast5 files and need to be basecalled using e.g. Guppy to obtain fastq files.
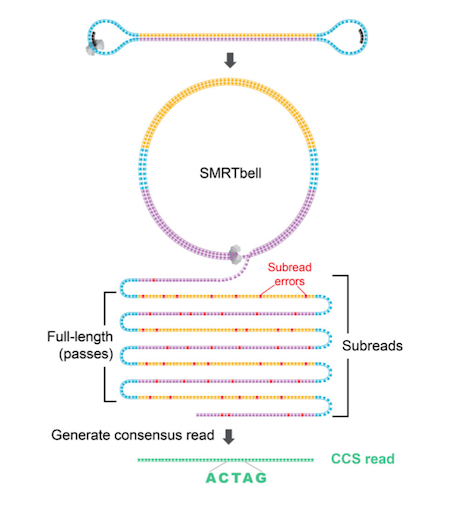
FASTQ has emerged as a common file format for sharing sequencing read data combining both the sequence and an associated per base quality score. For each sample you should have the corresponding FASTQ file (Illumina single-end (SR), PacBio and Nanopore data). For a Illumina paired-end (PE) run, you should have two FASTQ files (i.e. R1 and R2) per sample.
Nucleotide Archives¶
It is important that you share your data and maybe some other resources with the scientific community. Submit your sequencing raw data to any of the three data repositories. The European Nucleotide Archive (ENA), the GenBank / Sequence Read Archive (SRA) at NCBI, and the DNA DataBank of Japan (DDBJ) are all part of the International Nucleotide Sequence Database Collaboration and exchange data daily. There are other non-profit resource collection such as e.g. DYRAD to promote data publishing, curation, and preservation of all data types.
- International Nucleotide Sequence Database Collaboration
- European Nucleotide Archive (ENA)
- ENA: Guidelines and Tutorials
- DRYAD
Challenges¶
For the following challenges, I assume you are familiar with (NCBI) Blast searches. There are NCBI blast tutorials and NCBI webinars available to get started or to learn more.
First, we generate two 29nt-long random DNA sequences with a GC content of 50% and 20%. For this purpose you could use either the Random DNA generator provided by the Maduro lab at UCR or simply do it by hand.
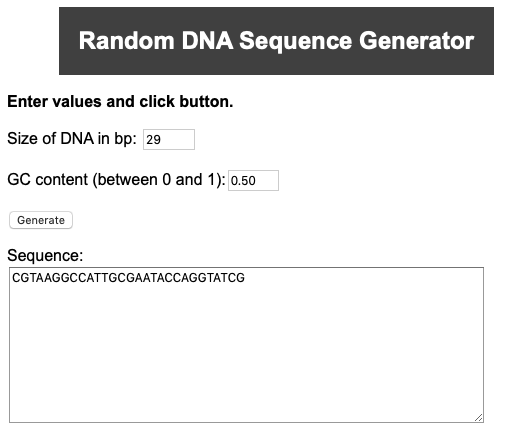
Possible sequence example:
>Seq1_GC50 CGTAAGGCCATTGCGAATACCAGGTATCG >Seq2_GC20 AAACTGTTAAAAAATCGTGTCTTTACAAT
Challenge #1: What is the text-based format representing the two nucleotide sequences called?
Solution #1
This is a sequence in FASTA format. Files containing nucleotide sequences carry the suffix fa (seq.fa) or fasta (seq.fasta). If the sequences are aligned the file ending is afa (alignment.afa). See help below to read more about sequence fasta format.
Challenge #2: How many possibilities are there to build a 29nt long sequences? In other words, what is the likelihood that we create the exact same sequences twice (with the same CG content)?
Solution #2
We have 4 possibilities (nucleotides) for each of the 29 sites, resulting in 4^29 = 2.88e17 possible nucleotide sequences. The likelihood is pretty slim!
Add the following "unknown" sequence to the previous two randomly generated sequences.
>Seq3_MysterySequence AATGATACGGCGACCACCGAGATCTACAC
Challenge #3: What is the GC content of the unknown mystery sequences?
Solution #3
It is ~52% (15/29) and similar to random sequence #1. You could also use comseq from the EMBOSS package with word size 1 to calcualte nucleotides frequencies. See help below for more information.
Next, we search the largest available nucleotide sequence collection to look for similar known sequences. For this purpose, we blast all three sequences against the NCBI nt BLAST-Database. What are your expectations?
My expectations are ...
Seq1: It is a well balanced (GC conten 0.5) random sequence and therfore we expect no perfect hits. The sequence is, however, short and the database is rather large and one-hit wonders cannot be fully excluded. Seq2: There is a good chance that we get some good hits because of the lower complexity. Complexity is an important factor in alinments. Seq3: Not sure. It seems that random blast hits depend on sequence length and complexit (among other factors). If it is a radom sequences, I would also expect no prefect hits similar to radom sequence #1. If there are multiple hits the sequences might not be random at all.
Interpret your Blast results carefully and compare it with your expectations. Do you have any idea what the unknown sequence #3 could be?
Blast results for sequences #3?
The blast result shows multiple perfect hits (100% similarity and 100% coverage). According to the blast results the unknown sequence could be a barn owl, a brown snake, a carp, or a bacterium and even a virus.
Do you have any good explanation for the blast results? Do you agree that it is not a bonanza but something real? Could it be an ultra-conserved genome element (UCE) or is it an artefacts? Maybe the internet can help? The sequence is short enough to be used in a Google search.
What is sequences #3?
It is not an UCE but more likely the result of careless NGS data preparation. The sequences is a perfect match with oligonucleotides used in Illumina (Nextera) library preparation. It is part of an adaptor-sequences you add to your target DNA fragments in order to sequence it. NGS quality reports should indicate adaptor contaminations and there are many tools freely available to remove such artificially introduced sequences prior to assembly.
Have a look at the Illumina adaptor sequences (link below) and blast a few more. You will see that the adaptor-contamination problem is widespread.
Tip
For the correct data preparation and analysis of your NGS data, it is important to know the detail about sample preparation and the sequence technology used. The more you know, the better you understand your data and possible artefacts.