SNPs

Introduction Notes¶
⬇︎ SNPs|caller
Variant types¶
Below you find the most common used variant types. For most downstream anaylsis only SNPs can be used.
| Type | Ref | Alt |
|---|---|---|
| SNP | A | T |
| INDEL | A | AT |
| MNP | AT | GC |
| CLUMPED | ATTTT | GTTTC |
Flagstat-output¶
| Flags | What does that mean? |
|---|---|
| total | Total reads. If part of the reads can be mapped on different location, the read will be duplicated. If you have many more reads than in your fastq file then you have a problem with the genome |
| mapped | All mapped reads |
| paired in sequencing | R1 and R2 could be mapped |
| properly paired | R1 and R2 could be mapped within insert range |
| with itself and mate mapped | R1 and R2 could be mapped also outside insert range |
| singletons | Either R1 or R2 could be mapped |
| mate mapped to a different chr | R1 or R2 could be mapped on different chromosomes |
Most of the reads should be properly paired. If not there is a problem with the reference used. If you do a mapping quality filtering, you need to extract the total number of reads from the unfiltered bam file.
Challenges¶
Post your comments, questions and ideas directly in the Google group.
Multiple sequence alignments (MSA)¶
In our first challenge we will perform a multiple sequence alignments of different SARS-CoV-2 strains.
There are many tool available to align multiple fasta sequences. We are going to use a webtool as there are only few sequences and the genome of the virus is pretty small.
Download the fasta file using this link here.
(1) How many strains do we have?
Suggestion
grep ">" -c SARS-CoV-2.fasta or count ">" in your editor
We are going to use mafft which runs at EMBL. Just use all default settings and upload the fasta file.
(2) Have a look at the alignment. What is the difference between an alignment and a fasta file?
(3) Have a look at the Neighbour-joining tree? What do you think about the topology?
Variants¶
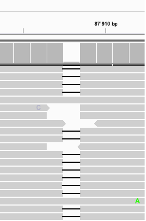
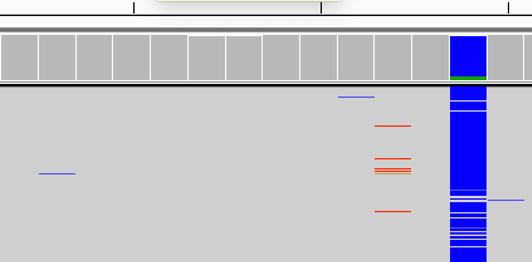
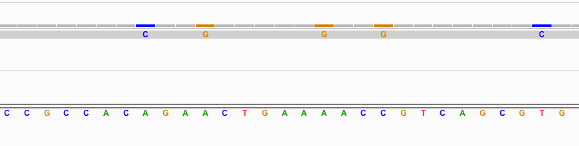
Find below some putative SNPs/variants. Is it a SNP or not and why?
Suggestion
A clear SNP as sufficient forward and reverse reads support it.
Suggestion
Could be an indel but it is not well supported.
Suggestion
Not a clear SNP as the alternative allele of the SNP is not well supported.
Suggestion
Not clear as the SNPs are supported by only 1 read.
Variant calling¶
For our next challenge we going to use a reduced data set containing different strains of the parasite Crithidia bombi infecting bumblebees. Find more information here.

We have 4 samples ERR3418932, ERR341896, ERR3418937 and ERR3418936 form different strains. To speed up the analysis we are going to use only 1 scaffold of the Crithidia bombi genome. Find more information about the genome here.
Login to our server.
ssh studentX@gdcsrv2.ethz.ch
Generate a new working folder, open it, download the data and open the tar file.
mkdir SNPs cd SNPs wget https://www.gdc-docs.ethz.ch/GeneticDiversityAnalysis/GDA20/data/SNP.tar.gz tar xvzf SNP.tar.gz ls
In your folder you will find the following files.
raw reads (e.g. ERR3418932_1.fq.gz and ERR3418932_2.fq.gz) reference genome (Ref_C_bombi.fasta) annotation e.g. where the genes are located (Ref_C_bombi.gff)
The aim is to first map the reads against the genome, do a variant calling, filter the variants and then check out manually some of the SNPs.
Mapping¶
First, we need to index the reference.
bwa index Ref_C_bombi.fasta
In order to make our code easier to read (e.g. more reproducible) we define variables.
sample=ERR3418937 Ref=Ref_C_bombi.fasta
We map reads to the reference using the default settings of bwa mem, add a read group and output a bam file.
bwa mem -R "@RG\tID:${sample}\tSM:${sample}\tPL:Illumina" ${Ref} ${sample}_1.fq.gz ${sample}_2.fq.gz |samtools view -b - > ${sample}.raw.bam
Now, we need to sort bam file.
samtools sort ${sample}.raw.bam -o ${sample}.sort.bam
(1) Check out the mapping statistics. How many reads could you map?
samtools flagstat ${sample}.sort.bam
Now, we remove reads with mapping quality lower than 10 to get rid of ambigous alignments.
samtools view -bhq 10 ${sample}.sort.bam > ${sample}.sort.Q10.bam
To account possible PCR duplicates we remove them and index the final bam file.
samtools rmdup ${sample}.sort.Q10.bam ${sample}.sort.Q10.nodup.bam
samtools index ${sample}.sort.Q10.nodup.bam
(2) Check again the mapping statistics to find out how many reads could have been mapped? What is the mapping rate?
To keep our folder tidy, let's delete the intermediate files.
rm ${sample}.raw.bam ${sample}.sort.Q10.bam
As we have now 3 more samples to map we are going to use bash loop to process all the samples in one command.
First, we generate a sample list and remove the endings.
ls *_1.fq.gz|sed 's/_1.fq.gz//' > sample.list
Then we use a loop to map all the samples in sample.list
#!/bin/bash ##define variable Ref=Ref_C_bombi.fasta ##get sample list ##ls *_1.fq.gz|sed 's/_1.fq.gz//' > sample.list ##bash loop to map reads against the genome for sample in $(cat samples.list) do bwa mem -R "@RG\tID:${sample}\tSM:${sample}\tPL:Illumina" ${Ref} ${sample}_1.fq.gz ${sample}_2.fq.gz \ |samtools view -b - > ${sample}.raw.bam samtools sort ${sample}.raw.bam -o ${sample}.sort.bam samtools flagstat ${sample}.sort.bam > ${sample}.sort.stats samtools view -hbq 10 ${sample}.sort.bam > ${sample}.sort.Q10.bam samtools rmdup ${sample}.sort.Q10.bam ${sample}.sort.Q10.nodup.bam samtools index ${sample}.sort.Q10.nodup.bam samtools flagstat ${sample}.sort.Q10.nodup.bam > ${sample}.sort.Q10.nodup.stats rm ${sample}.sort.bam ${sample}.sort.Q10.bam done
Let's copy the script above in a text file and call it mapping.sh. Add comments.
We need then to make the file executable.
chomd +x mapping.sh
And execute our bash script.
./mapping.sh
(3) Have a look at the mapping statistics. What do you think about the mapping rates across the samples (check out .sort.stats and .sort.Q10.nodup.stats)?
Suggestion
| Sample | total | mapped | % | After filtering | % |
|------------|--------|--------|------|-----------------|------|
| ERR3418932 | 130943 | 122767 | 0.94 | 108753 | 0.83 |
| ERR3418936 | 126253 | 123067 | 0.97 | 110931 | 0.88 |
| ERR3418937 | 98889 | 77652 | 0.79 | 67921 | 0.69 |
| ERR3418961 | 106936 | 4798 | 0.04 | 3082 | 0.03 |
grep "in total (" *.sort.stats
grep "mapped (" *.sort.stats
grep "mapped (" *.Q10.nodup.stats
SNP calling¶
First, we need to generate a list with all bam files as we like to do a joint calling.
ls -1 *.sort.Q10.nodup.bam > bamfiles.txt
Let's now run FreeBayes using the default settings.
freebayes -p 2 -L bamfiles.txt -v raw.vcf -f ${Ref}
(1) How many SNPs and other variants do you have in total?
We count the number of lines but exclude lines starting with #.
cat TotalRawSNPs.vcf| grep -v '^#' | wc -l
(2) Have a look at the SNP table. Try to understand the vcf file format. What are the genotypes of the 4 samples at scaffold3_8_ref0000002_ref0000084 and position 100.
Suggestions
scaffold3_8_ref0000002_ref0000084 100
ERR3418932 AA
ERR3418961 NA
ERR3418937 TT
ERR3418936 AA
SNP filtering¶
In order to filter false positive SNPs we are going to apply some hard filters. The filters peresented here is only a subset. When we are working with RAD SNPs, we will see more.
Set sites with less than 3 reads to NA and keep only sites with a minimum mapping quality of 20.
vcftools --vcf raw.vcf --minQ 20 --minDP 3 --recode --recode-INFO-all --out raw_Q20_minDP3
(1) How many variants are you retaining?
Remove sites with more than 5% missing sites.
vcftools --vcf raw_Q20_minDP3.recode.vcf --max-missing 0.95 --recode --recode-INFO-all --out raw_Q20_minDP3_NA95
(2) How many variants do you retain?
Let's decompose haplotypes into single SNPs.
vcfallelicprimitives -g -k raw_Q20_minDP3_NA95.recode.vcf > raw_Q20_minDP3_NA95_SNPs.vcf
Finally, we only keep biallelic sites and remove indels.
vcftools --vcf raw_Q20_minDP3_NA95_SNPs.vcf --min-alleles 1 --max-alleles 2 --remove-indels --out raw_Q20_minDP3_NA95_SNPs_noIndels
(3) What is the final numbers of SNPs in your table?
Manual inspection of the SNPs¶
First, we need to index the fasta file.
samtools faidx Ref_C_bombi.fasta
Download and install IGV on your own computer use the link here.
Transfer the alignments (bams), the raw and the filtered SNP table (vcf), the reference (fasta and the Index) and the annotation (gff) file.
Open IGV and create a new genome.
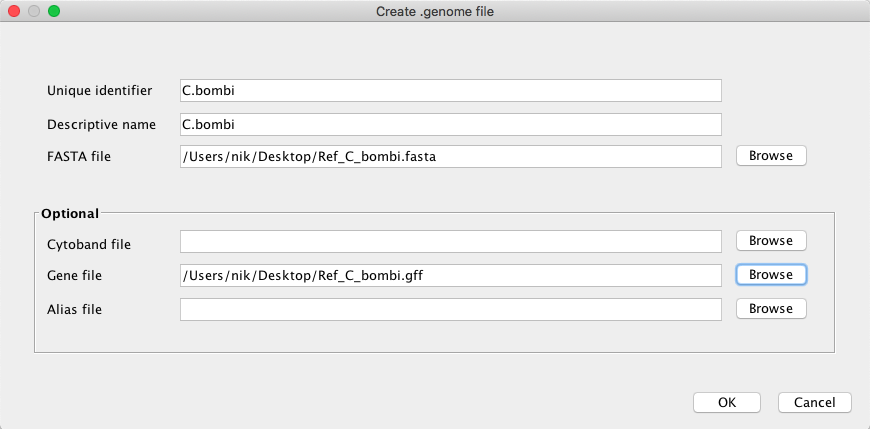
Drag and drop the bam and vcf files. Can you verify some of the SNPs manually? Do you see differences between the filter and the raw vcf? Make some print screens and post them.
Additional Information¶
File formats¶
Alignments¶
- Local versus global alignment
- Global Alignments
- Alignement tool
- BLAST tutorial
- NGS alignements
- Decoding SAM flags